A Comprehensive Guide to Single-Cell RNA Sequencing Data Analysis: From Foundations to Clinical Applications
This article provides a comprehensive guide to single-cell RNA sequencing (scRNA-seq) data analysis, tailored for researchers, scientists, and drug development professionals.
A Comprehensive Guide to Single-Cell RNA Sequencing Data Analysis: From Foundations to Clinical Applications
Abstract
This article provides a comprehensive guide to single-cell RNA sequencing (scRNA-seq) data analysis, tailored for researchers, scientists, and drug development professionals. It covers the foundational principles of scRNA-seq workflows, including quality control, normalization, and clustering. The guide then explores advanced methodological applications, such as machine learning integration and analysis in drug discovery. It addresses critical troubleshooting and optimization strategies for technical noise and data integration challenges. Finally, it outlines robust validation and comparative analysis techniques to ensure biological relevance and reproducibility. This resource synthesizes current best practices and emerging trends to empower robust and insightful single-cell research.
Mastering the scRNA-seq Pipeline: Essential Steps from Raw Data to Cell Clustering
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the characterization of gene expression at the resolution of individual cells. This technology has progressed substantially since its inception, with two primary methodologies emerging as dominant: plate-based and droplet-based approaches [1]. The selection between these platforms is a critical initial step in experimental design, impacting everything from cost and throughput to data quality and biological insights [2]. Within the broader context of a thesis on single-cell RNA sequencing data analysis, understanding the foundational technologies that generate the data is paramount, as the choice of wet-lab methodology directly influences downstream analytical strategies and computational requirements [3].
This document provides a comprehensive comparison of plate-based and droplet-based scRNA-seq methods, offering detailed protocols and application notes to guide researchers, scientists, and drug development professionals in selecting and implementing the most appropriate technology for their research objectives.
Technology Comparison: Plate-Based vs. Droplet-Based Methods
The core distinction between plate-based and droplet-based scRNA-seq methods lies in their mechanism for isolating individual cells and barcoding their transcripts [1]. The following table summarizes the key characteristics of each approach, which are crucial for experimental planning.
Table 1: Comparative analysis of plate-based and droplet-based scRNA-seq technologies
| Feature | Plate-Based scRNA-seq | Droplet-Based scRNA-seq |
|---|---|---|
| Throughput | Lower (although combinatorial indexing improves scalability) [1] | Highest [1] |
| Cost per Cell | Highest, due to greater reagent consumption [1] | Lowest, due to microfluidics miniaturization [1] |
| Sensitivity | Highest [1] | Lower than plate-based [1] |
| Workflow | Flexible but labor-intensive (involves manual cell sorting and pipetting) [1] | Highly automated, but requires expensive microfluidics equipment [1] |
| Cell Isolation | Fluorescence-activated cell sorting (FACS) into multi-well plates or combinatorial indexing [1] | Microfluidic encapsulation of single cells in droplets [1] [4] |
| Best For | Smaller-scale, in-depth studies; full-length transcript analysis [1] [5] | Large-scale studies, such as cell atlas projects and complex tissue analysis [1] [4] |
| Multiplet Rate | Controlled during cell sorting | Typically <5% when following optimal loading concentrations [4] |
The following workflow diagram illustrates the fundamental procedural differences between these two core technologies.
Figure 1: Core Workflows for scRNA-seq Methods. This diagram contrasts the key steps in plate-based (green) and droplet-based (blue) scRNA-seq protocols, highlighting the initial cell isolation and barcoding strategies.
Detailed Experimental Protocols
Core Protocol for Droplet-Based scRNA-seq (10x Genomics Chromium)
The droplet-based method is designed for high-throughput analysis, enabling the profiling of thousands to millions of cells in a single experiment [4].
Workflow Details:
- Single-Cell Suspension Preparation: Begin by preparing a high-quality single-cell suspension. Optimize cell concentration to 700–1,200 cells/μL and ensure viability exceeds 85% to minimize ambient RNA [4]. Maintain cells on ice to arrest metabolic activity and reduce stress-induced gene expression [2].
- Microfluidic Partitioning and Barcoding: Combine the cell suspension with barcoded gel beads and partition oil on a microfluidic chip. This generates monodisperse, nanoliter-scale Gel Bead-in-Emulsions (GEMs) [4]. Each GEM ideally contains a single cell and a single bead. The beads are coated with oligonucleotides containing unique molecular identifiers (UMIs), cell barcodes, and poly(dT) sequences for mRNA capture [1] [4].
- Within-Droplet Reactions: Inside each droplet, cells are lysed, releasing mRNA. The poly(A) tails of mRNA transcripts hybridize to the poly(dT) primers on the beads. Reverse transcription then occurs, producing barcoded cDNA molecules, with each molecule tagged with the same cell barcode and a unique UMI to correct for amplification biases [4].
- Post-Processing and Library Construction: The emulsion is broken, and the barcoded cDNA from all droplets is pooled. The cDNA is then amplified via PCR, and sequencing adapters are ligated to create the final library [1]. After quality control, the library is sequenced on a high-throughput platform.
Core Protocol for Plate-Based scRNA-seq (SMART-Seq2)
Plate-based methods, such as SMART-Seq2, prioritize sensitivity and full-length transcript coverage, making them ideal for focused studies [5].
Workflow Details:
- Cell Sorting and Lysis: Use fluorescence-activated cell sorting (FACS) to isolate individual cells into the wells of a 96- or 384-well plate containing lysis buffer [1]. The plate is then centrifuged to ensure the cell is immersed in the buffer.
- Reverse Transcription and cDNA Amplification: Lyse the cells to release RNA. Reverse transcription is initiated using an oligo(dT) primer and a template-switching oligo (TSO), which allows for the synthesis of full-length cDNA with defined ends. The cDNA is then amplified via PCR to generate sufficient material for library construction [5].
- Library Preparation and Sequencing: The amplified cDNA from each well is fragmented, and sequencing adapters are ligated. In modern protocols like SMART-seq3, cell-specific barcodes can be incorporated during library preparation, allowing for the pooling of multiple cells before sequencing. However, earlier protocols required separate library preparation for each cell [1]. The libraries are then sequenced.
Protocol for Single-Nucleus RNA-seq from Frozen Tissue
For tissues that are difficult to dissociate or uniquely valuable, such as archived clinical samples, single-nucleus RNA-seq (snRNA-seq) from frozen tissue is a robust alternative [6]. The following diagram outlines a simplified and optimized preparation method.
Figure 2: Single-Nucleus Isolation from Frozen Tissue. This protocol is optimized for long-term frozen brain tumor tissues but can be adapted for other challenging samples [6].
Key Considerations:
- This protocol is fast (under 30 minutes), low-cost, and yields intact nuclei with minimal debris [6].
- The resulting nuclei are compatible with both droplet-based (e.g., 10X Genomics Chromium, Drop-seq) and plate-based (e.g., Fluidigm C1) platforms [6].
- Two washes are recommended for low starting material to maximize yield, while three washes provide a debris-free supernatant for higher-quality input [6].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful scRNA-seq experiments rely on a suite of specialized reagents and instruments. The following table details key solutions and their functions in the experimental workflow.
Table 2: Key Research Reagent Solutions for scRNA-seq
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Barcoded Gel Beads | Microbeads containing millions of oligonucleotides with cell barcodes, UMIs, and poly(dT) for mRNA capture and labeling within droplets [4]. | 10x Genomics Chromium systems; essential for droplet-based multiplexing. |
| Template-Switching Oligo (TSO) | An oligonucleotide that enables the synthesis of full-length cDNA with defined ends during reverse transcription, independent of poly(A) tails [4]. | Used in SMART-Seq2 and other plate-based protocols for superior transcript coverage. |
| Unique Molecular Identifiers (UMIs) | Short random nucleotide sequences added to each transcript during reverse transcription; used to correct for PCR amplification bias and quantitatively count original mRNA molecules [4]. | Standard in droplet-based methods (e.g., 10x, Drop-seq) and present in modern plate-based methods (e.g., SMART-seq3). |
| Commercial Enzyme Cocktails | Pre-optimized mixtures of enzymes for rapid and reproducible tissue dissociation into single-cell suspensions. | Miltenyi Biotec kits and the gentleMACS Dissociator for standardized sample prep [2]. |
| Density Gradient Media | Solutions like Ficoll or Optiprep used to separate viable cells/nuclei from debris and dead cells via centrifugation [2] [6]. | Cleaning up peripheral blood mononuclear cells (PBMCs) or nuclei isolated from brain tissue [2]. |
| Fixation Reagents | Chemicals (e.g., paraformaldehyde) that preserve cells or nuclei, allowing samples to be stored and batched for processing to minimize technical variability [2]. | Critical for large-scale projects, time-course experiments, and clinical samples with unpredictable arrival times. |
Critical Factors in Experimental Design
Sample Preparation and Quality Control
The foundation of a successful scRNA-seq experiment is a high-quality single-cell or single-nucleus suspension.
- Cells vs. Nuclei: The decision to sequence whole cells or nuclei depends on the sample type and research question. Nuclei are preferable for difficult-to-dissociate tissues (e.g., brain, fibrous tumors), frozen archived samples, or when working with very large cells that exceed the size limit of droplet-based systems [2] [6].
- Viability and Debris: Aim for sample viability between 70% and 90%. Minimize cell clumping and debris (<5% aggregation) by filtering the suspension and using calcium/magnesium-free media. Accurate cell counting is critical before loading onto any platform [2].
- Fresh vs. Fixed: While fresh processing is ideal, fixation allows for sample storage and batching, which is invaluable for clinical settings and large-scale projects. Fixed samples help control for batch effects and provide logistical flexibility [2].
Replication and Sample Size
Adequate replication is essential for robust and statistically sound conclusions.
- Biological vs. Technical Replicates: Biological replicates (e.g., cells from different donors) capture inherent biological variability and are necessary to verify reproducibility. Technical replicates (sub-samples from the same biological sample) measure noise from the protocol or equipment [2].
- Cell Number: The required number of cells depends on the biological question. Pilot studies or experimental planning tools like the Single Cell Experimental Planner can help determine the necessary scale. Technologies like combinatorial barcoding (e.g., Parse Biosciences' Evercode) allow for the processing of up to 1 million cells without the need for physical partitioning, offering great scalability [1] [2].
The choice between plate-based and droplet-based scRNA-seq methods is a fundamental strategic decision that shapes the entire research pipeline. Plate-based methods offer high sensitivity and are ideal for focused studies where detailed characterization of individual cells is paramount. In contrast, droplet-based platforms provide unparalleled scalability for large-scale profiling of complex tissues and discovery of rare cell types. The ongoing development of integrated multi-omics approaches, combined with improved bioinformatic tools, continues to expand the applications of scRNA-seq in fields like cancer research, reproductive medicine, and drug development [4]. By carefully considering the factors outlined in this document—throughput, cost, sensitivity, and sample requirements—researchers can select the most appropriate technology to generate high-quality data, thereby laying a solid foundation for meaningful biological insights and advancements in translational medicine.
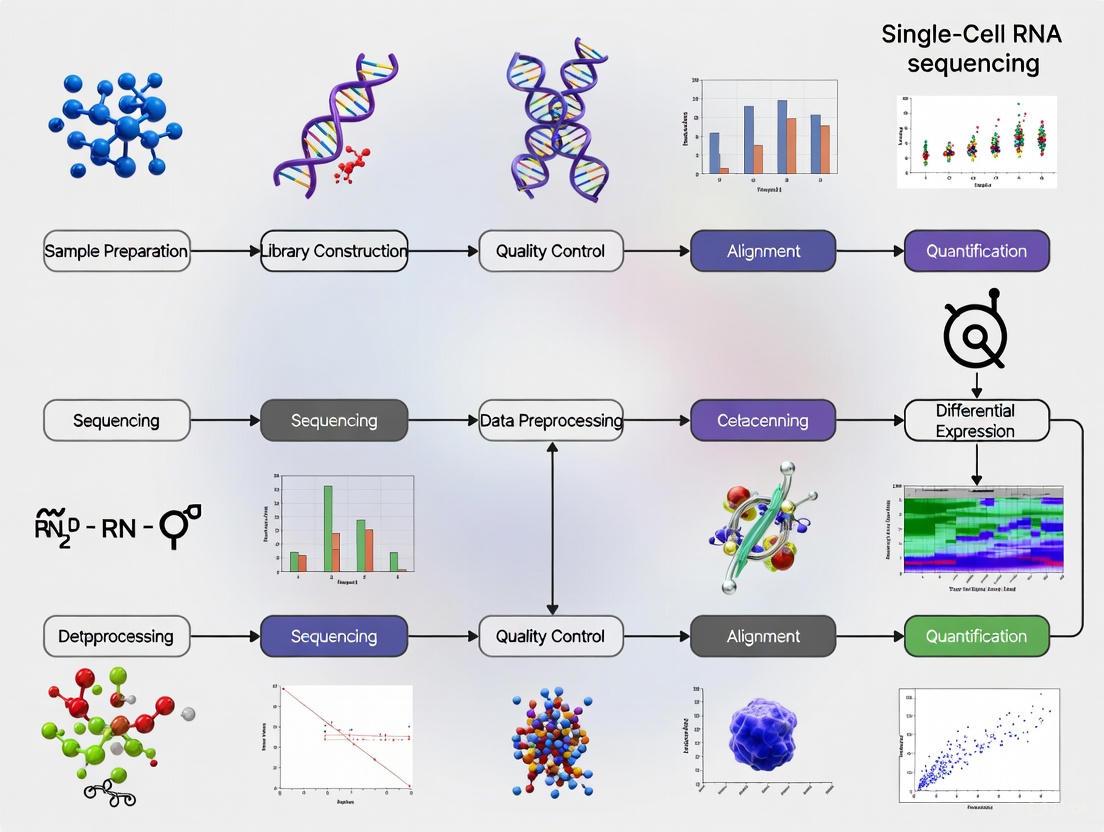
In single-cell RNA sequencing (scRNA-seq) research, the initial phase of raw data processing and quantification is a critical determinant of the validity of all subsequent biological interpretations. This foundational step transforms the billions of sequencing reads contained in FASTQ files into a structured gene expression count matrix, enabling the exploration of cellular heterogeneity at unprecedented resolution. The complexity of this process, involving meticulous quality control, genome alignment, and molecular counting, presents significant computational and methodological challenges. This application note delineates the established protocols and emerging best practices for this crucial conversion, framed within the rigorous context of academic research and drug development. A standardized approach ensures data integrity, minimizes technical artifacts, and provides a reliable foundation for uncovering novel biological insights and therapeutic targets, making proficiency in this initial stage indispensable for researchers and scientists in the field.
The journey from raw sequencing data to a quantitative gene expression matrix involves a series of computationally intensive and methodologically nuanced steps. The process begins with sequencing core facilities typically providing FASTQ files, which are text-based files storing nucleotide sequences and their corresponding quality scores for every read [7]. The primary objective of the initial processing pipeline is to generate a count matrix, where each row represents a gene, each column represents an individual cell, and each value contains the number of RNA molecules originating from a particular gene in a particular cell [7] [8].
Table 1: Key Components of a Processed Count Matrix
| Component | Format | Description |
|---|---|---|
| Sparse Matrix (MTX) | .mtx file |
A compact format storing the non-zero count data efficiently. |
| Cell Metadata | TSV/CSV file | Contains cellular barcode information identifying each cell. |
| Gene List | TSV/CSV file | Includes gene names, IDs, and other feature annotations. |
The following diagram illustrates the logical sequence and decision points in a standard scRNA-seq raw data processing workflow, from the acquisition of FASTQ files to the final quality-checked count matrix.
Detailed Methodologies and Protocols
FASTQ File Processing and Alignment
The first computational stage involves processing the raw FASTQ files. An initial quality control (QC) check is performed using tools like FastQC or MultiQC to visualize sequencing quality and validate information [7]. This step identifies potential issues such as adapter contamination, low-quality bases, or overrepresented sequences, which could compromise downstream analysis.
Following QC, reads must be aligned to a reference genome. This essential step maps each sequenced read to its genomic location of origin. Common open-source alignment tools include STAR and kallisto | bustools [7]. The choice of alignment tool can depend on factors such as accuracy, speed, and computational resources. For UMI-based protocols, a critical subsequent step is deduplication, where PCR-amplified copies of the same original mRNA molecule are identified based on their shared UMI and counted only once, thus moving from read counts to molecule counts and mitigating amplification bias [8].
Table 2: Comparison of Selected scRNA-seq Data Processing Pipelines
| Pipeline/Tool | Best For / Key Feature | Input | Primary Output |
|---|---|---|---|
| Cell Ranger | 10x Genomics data integration; widely adopted standard [9]. | FASTQ | Filtered count matrix |
| Parse Biosciences' Trailmaker | Processing data from Parse's combinatorial barcoding method [7]. | FASTQ | Count matrix |
| zUMIs | Flexible pipeline for various UMI-based protocols [8]. | FASTQ | Count matrix |
| SEQC | Handles data from sequence-based single-cell methods [8]. | FASTQ | Count matrix |
| nf-core/scrnaseq | Community-built, portable Nextflow pipeline [7]. | FASTQ | Count matrix |
Post-Processing Quality Control and Filtering
Once a count matrix is generated, rigorous filtering is required to ensure that only high-quality data is retained. This involves distinguishing genuine cells from artifacts [7].
- Removing Background RNA: In droplet-based methods, droplets without cells can contain free-floating mRNA that is barcoded, creating background noise. Classifier filters or knee plots can distinguish barcodes corresponding to real cells from those associated with background. A common threshold is to set a minimum of 200-500 transcripts per cell, though this is adjustable based on biological context [7].
- Identifying Dead or Dying Cells: Cells with compromised membranes have a characteristically high fraction of reads mapping to mitochondrial genes. A common filtering threshold is 10-20% mitochondrial read fraction, though this varies by cell type. For example, nuclei should have virtually no mitochondrial reads [7] [8].
- Identifying and Removing Doublets: Doublets occur when two or more cells are tagged with the same barcode, potentially creating artifactual cell states. Bioinformatics tools like Scrublet (for Python) and DoubletFinder (for R) are designed to identify and remove doublets by comparing cell expression profiles to artificially generated doublets [7].
Tools like SoupX and CellBender can be applied to computationally estimate and remove ambient RNA contamination, a common issue in droplet-based datasets [7].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
The successful execution of a scRNA-seq study relies on a combination of wet-lab reagents and dry-lab computational resources.
Table 3: Essential Research Reagent and Computational Solutions
| Category / Item | Function / Description |
|---|---|
| Library Prep Kits | |
| 10x Chromium Single Cell 3' Kit | Integrated solution for droplet-based single-cell partitioning, barcoding, and library prep. |
| Parse Biosciences Single-Cell Kit | Uses combinatorial in-situ barcoding in a plate-based format [7]. |
| Critical Computational Tools | |
| Cell Ranger (10x Genomics) | End-to-end analysis pipeline for demultiplexing, alignment, and counting from FASTQ files [9]. |
| Seurat / Scanpy | Comprehensive R/Python environments for downstream analysis after count matrix generation [7] [8]. |
| Reference Genomes | |
| GENCODE Human (GRCh38) | High-quality, annotated reference genome for accurate read alignment. |
| Quality Control Tools | |
| FastQC / MultiQC | Provide initial assessment of sequencing run quality [7]. |
| CellBender | Removes technical artifacts and background RNA from count matrices [9]. |
Downstream Analysis and Advanced Computational Tools
Upon obtaining a high-quality count matrix, researchers embark on the biological discovery phase. This involves data normalization to account for differences in sequencing depth between cells, often followed by log-transformation to stabilize variance [7]. Dimensionality reduction techniques like PCA and UMAP are then applied to visualize cells in a low-dimensional space, facilitating the identification of clusters representing distinct cell types or states [7] [10].
Newer model-based dimensionality reduction methods, such as scGBM, are being developed to directly model count data and better capture biological variability while quantifying uncertainty in the low-dimensional representation [11]. For specialized applications like single-cell CRISPR screening (e.g., Perturb-seq), integrated analysis pipelines such as MUSIC are available to quantitatively estimate the effect of genetic perturbations on single-cell gene expression profiles [12].
A modern ecosystem of cloud-based platforms, including Nygen and BBrowserX, now offers user-friendly, no-code interfaces for performing these downstream analyses, making scRNA-seq data interpretation more accessible to non-bioinformaticians [10].
Quality control (QC) represents a critical first step in single-cell RNA sequencing (scRNA-seq) data analysis, serving as the foundation for all subsequent biological interpretations. Within the broader thesis of scRNA-seq data analysis research, rigorous QC procedures are indispensable for distinguishing technical artifacts from genuine biological signals [13]. The primary goals of QC include generating metrics to assess sample quality and removing poor-quality data that could confound analysis and interpretation [14]. Without standardized QC practices, researchers risk deriving erroneous conclusions from clusters of stressed, dying, or multiple cells mistakenly identified as distinct cell populations [13]. This application note provides a comprehensive framework for implementing rigorous QC protocols, encompassing the filtering of cells, genes, and detection of doublets, specifically tailored for researchers, scientists, and drug development professionals working with scRNA-seq data.
Key Quality Control Metrics and Their Biological Significance
Standard QC Metrics for Cell Filtering
The evaluation of single-cell data quality relies on several key metrics that help identify and remove problematic barcodes. Table 1 summarizes the primary QC metrics, their biological or technical significance, and common filtering approaches.
Table 1: Essential QC Metrics for Single-Cell RNA-Seq Data
| QC Metric | Technical/Biological Significance | Common Filtering Approaches | Special Considerations |
|---|---|---|---|
| UMI Counts | Represents absolute number of observed transcripts; low counts may indicate empty droplets or damaged cells; high counts may indicate multiplets [14]. | Data-driven thresholds (3-5 times standard deviation from median); arbitrary cutoffs; Cell Ranger caps at 500 UMIs [14]. | Heterogeneous samples may require cell type-specific thresholds; neutrophils naturally have low RNA content [15] [14]. |
| Genes Detected | Number of unique genes detected per cell; correlates with UMI counts; extreme values indicate multiplets or empty droplets [14]. | Similar approach to UMI filtering; often applied in conjunction [14]. | Varies by cell type; filtering thresholds should account for biological heterogeneity [14]. |
| Mitochondrial Gene Percentage | Increased levels associated with stressed, apoptotic, or low-quality cells where cytoplasmic RNA has leaked out [16] [14]. | Typical thresholds: 5-25% depending on cell type; data-driven thresholds (3-5 times SD/MAD from median) [14]. | Cardiomyocytes and other metabolically active cells naturally high mt content; filtering may introduce bias [14]. |
| Doublet Score | Computational prediction of droplets containing multiple cells; creates hybrid expression profiles [13] [14]. | Thresholds on doublet scores from specialized tools; subjective and data-dependent [14]. | Essential for preventing misinterpretation of multiplets as novel cell types or transitional states [13]. |
Platform-Specific QC Considerations
The performance of QC metrics can vary significantly across experimental platforms. A 2025 comparative analysis of technologies from 10× Genomics, PARSE Biosciences, and Honeycomb Biotechnologies revealed notable differences in baseline quality metrics when profiling challenging cell types like neutrophils [15]. For instance, mitochondrial gene expression levels were generally low (0-8%) across most technologies but reached up to 25% in Chromium Single-Cell 3′ Gene Expression v.3.1, highlighting the importance of platform-aware threshold setting [15].
Droplet-based technologies present unique QC challenges distinct from plate-based methods. Specifically, the majority of droplets (>90%) in microfluidic devices do not contain an actual cell, necessitating specialized algorithms to distinguish empty droplets containing only ambient RNA from cell-containing droplets [13]. Technologies like Parse Biosciences' Evercode, which employs combinatorial barcoding, may demonstrate different quality metric distributions compared to droplet-based platforms, sometimes showing less distinct bimodal distributions in gene expression for complex samples [15].
Comprehensive QC Workflow and Experimental Protocols
Integrated QC Pipeline
The QC process should follow a systematic workflow that incorporates multiple complementary approaches to ensure comprehensive quality assessment. The following diagram illustrates the integrated QC pipeline for scRNA-seq data:
Protocol: Step-by-Step QC Implementation
3.2.1 Data Preprocessing and Empty Droplet Detection
Step 1: Raw Data Processing - Process raw FASTQ files using Cell Ranger or equivalent alignment and UMI counting pipelines [16]. The output will include a feature-barcode matrix containing both cell-containing and empty droplets.
Step 2: Empty Droplet Identification - Apply algorithms such as
barcodeRanksandEmptyDropsfrom theDropletUtilspackage to distinguish true cells from empty droplets [13] [14]. These methods work by ranking barcodes based on UMI counts and identifying the "knee" point in the log-log plot of rank against total counts, where barcodes below this point represent empty droplets [13].
3.2.2 Comprehensive QC Metric Calculation
- Step 3: Metric Computation - Calculate standard QC metrics including:
3.2.3 Doublet Detection and Ambient RNA Correction
Step 4: Computational Doublet Identification - Run doublet detection algorithms such as DoubletFinder or Scrublet, which generate artificial doublets and compare gene expression profiles of barcodes against these in silico doublets to calculate a doublet score [14]. The threshold for doublet filtering is data-dependent and should be determined by examining the distribution of doublet scores.
Step 5: Ambient RNA Correction - Apply tools like SoupX, DecontX, or CellBender to estimate and remove contamination from ambient RNA [14]. These algorithms model the background RNA profile and subtract its contribution from each cell's expression counts [13].
Protocol: Threshold Determination and Cell Filtering
3.3.1 Data Visualization for Threshold Setting
- Step 6: Quality Metric Visualization - Generate violin plots, box plots, or density plots to visualize the distribution of QC metrics (UMI counts, genes detected, mitochondrial percentage) across all cells [14]. This visualization helps identify appropriate filtering thresholds before proceeding with actual filtering.
3.3.2 Iterative Filtering Approach
Step 7: Application of Filters - Implement filtering decisions based on the established thresholds. As emphasized in best practices, "begin with permissive filtering approaches, and then revisit the filtering parameters if the downstream analysis results cannot be interpreted" [14].
Step 8: Quality Assessment - Regenerate visualization plots post-filtering to confirm the removal of outliers while preserving biologically relevant cell populations.
The following diagram illustrates the decision process for setting appropriate filtering thresholds:
Research Reagent Solutions
Table 2: Essential Research Reagents and Computational Tools for scRNA-seq QC
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| Chromium Next GEM Single Cell 3ʹ | Wet-bench Reagent | Single-cell library preparation | 10x Genomics platform; optimized for cell suspensions [17] |
| Chromium Nuclei Isolation Kit | Wet-bench Reagent | Nuclei isolation for snRNA-seq | Frozen samples; difficult-to-dissociate tissues [17] |
| Cell Ranger | Computational Pipeline | Raw data processing, alignment, UMI counting | 10x Genomics data; generates feature-barcode matrices [16] |
| SingleCellTK (SCTK-QC) | R/Bioconductor Package | Comprehensive QC metric calculation and visualization | Integrates multiple QC tools; user-friendly interface [13] |
| EmptyDrops | Algorithm | Empty droplet detection | Distinguishes cells from empty droplets in droplet-based data [13] [14] |
| DoubletFinder | Algorithm | Doublet/multiplet detection | Identifies droplets containing multiple cells [14] |
| SoupX | Algorithm | Ambient RNA correction | Removes background RNA contamination [16] [14] |
Specialized Considerations for Challenging Cell Types
Certain cell types present unique challenges for QC filtering and require specialized approaches. Neutrophils, for instance, contain naturally low levels of RNA compared to other blood cell types, which can lead to their inadvertent removal during standard QC filtering [15] [14]. A 2025 study demonstrated that applying a minimum threshold of 50 genes and 50 UMIs was necessary to ensure neutrophil inclusion in downstream analyses [15]. Similarly, cardiomyocytes and other metabolically active cells may exhibit naturally high mitochondrial content, necessitating adjusted thresholds to prevent the loss of biologically intact cells [14].
The choice between single-cell and single-nuclei RNA sequencing also impacts QC procedures. snRNA-seq, while applicable to frozen biobanked samples, primarily captures nuclear transcripts, resulting in different gene detection profiles compared to scRNA-seq [17]. Research has shown that cell type proportion differences between annotation methods were larger for snRNA-seq than scRNA-seq, highlighting the need for tailored QC and annotation strategies for nuclear data [17].
Rigorous quality control represents a non-negotiable foundation for robust scRNA-seq data analysis. By implementing the comprehensive QC framework outlined in this application note—encompassing standardized metric calculation, platform-aware threshold setting, and specialized handling of challenging cell types—researchers can significantly enhance the reliability of their biological conclusions. The integrated approach combining multiple complementary QC methods provides a robust defense against technical artifacts that could otherwise compromise data interpretation. As single-cell technologies continue to evolve and find expanded applications in drug development and clinical biomarker discovery [15], establishing and maintaining rigorous QC protocols will remain essential for generating biologically meaningful and reproducible results.
Data Normalization and Feature Selection for Dimensionality Reduction
Within the broader context of single-cell RNA sequencing (scRNA-seq) data analysis research, the steps of data normalization and feature selection are critical prerequisites for effective dimensionality reduction. The high-dimensional, sparse, and noisy nature of scRNA-seq data, characterized by an abundance of zero counts and technical variability from sources like sequencing depth and capture efficiency, necessitates robust preprocessing pipelines [18]. The curse of dimensionality further underscores that higher-dimensional data often contains more noise and redundancy, which does not necessarily benefit downstream analysis [19]. The choices made during normalization and feature selection have a profound and direct impact on the performance of subsequent dimensionality reduction techniques, such as PCA, UMAP, and t-SNE, which are essential for visualizing cellular heterogeneity, identifying novel cell types, and tracing developmental lineages [20] [21]. Consequently, this application note provides detailed protocols and a comparative analysis of current methods to guide researchers and drug development professionals in constructing reliable and interpretable analysis workflows.
Comparative Analysis of Methods
Data Normalization Methods
Normalization aims to remove technical variation while preserving biological variation, making gene counts comparable within and between cells [18] [22]. The table below summarizes commonly used normalization methods, their underlying models, and key features.
Table 1: Comparison of Single-Cell RNA-Seq Data Normalization Methods
| Method | Model/Approach | Key Features | Implementation |
|---|---|---|---|
| Log-Norm | Global scaling + log transformation | Divides counts by total per cell, scales (e.g., 10,000), adds pseudocount (e.g., 1), and log-transforms. Simple and widely used. | Seurat (NormalizeData), Scanpy (normalize_total, log1p) |
| SCTransform | Regularized Negative Binomial GLM | Models counts with sequencing depth as covariate; outputs Pearson residuals that are independent of sequencing depth. | R (Seurat) |
| Scran | Pooling and linear decomposition | Pools cells to sum counts, normalizes against a reference pseudo-cell, and solves linear system for cell-specific size factors. | R (scran) |
| BASiCS | Bayesian Hierarchical Model | Uses spike-in genes or technical replicates to jointly model technical and biological variation. | R (BASiCS) |
| SCnorm | Quantile Regression | Groups genes by dependence on sequencing depth; estimates and applies group-specific scale factors. | R (SCnorm) |
| Linnorm | Linear model and transformation | Optimizes a transformation parameter to achieve homoscedasticity and normality before linear model fitting. | R (Linnorm) |
| PsiNorm | Pareto Type I Distribution | Uses the shape parameter of a Pareto distribution as a multiplicative normalization factor; highly scalable. | R (PsiNorm) |
Feature Selection Methods
Feature selection reduces dimensionality by identifying a subset of informative genes, which is crucial for mitigating noise and enhancing the performance of downstream integration and clustering [23] [24]. The following table benchmarks different feature selection approaches.
Table 2: Comparison of Feature Selection Methods for scRNA-seq Data
| Method | Principle | Use Case | Considerations |
|---|---|---|---|
| Highly Variable Genes (HVG) | Selects genes with high variance-to-mean ratio. | Standard practice for reference atlas construction and integration [23]. | Sensitive to normalization and pseudocount choice [24]. |
| Deviance-based | Ranks genes by binomial deviance from a constant expression null model. | Effective for capturing biological heterogeneity; works on raw counts [24]. | Computed in closed form; implemented in the scry R package. |
| Highly Expressed Genes | Selects genes with the highest average expression. | A simple, traditional approach. | May select ubiquitously expressed housekeeping genes. |
| Stably Expressed Genes (e.g., scSEGIndex) | Selects genes with minimal biological variability. | Serves as a negative control in benchmarking [23]. | Not suitable for identifying biologically variable features. |
Experimental Protocols
Protocol I: Data Normalization using SCTransform
SCTransform is a robust method that effectively normalizes data and stabilizes variance in a single step [22].
Detailed Methodology:
- Input Data: Begin with a raw UMI count matrix ((X_{\text{raw}})) where rows are genes and columns are cells.
- Model Fitting: For each gene, a regularized Negative Binomial generalized linear model is fitted: ( \text{UMI Count} \sim \log(\text{Total UMI per Cell}) ) This model accounts for the relationship between gene expression and sequencing depth.
- Parameter Regularization: Model parameters (intercept, slope, dispersion) are regularized based on their relationship with the gene's mean expression across cells. This step prevents overfitting, which is critical for noisy scRNA-seq data.
- Residual Calculation: The regularized parameters are used to compute Pearson residuals: ( \text{Residual} = \frac{\text{Observed Count} - \text{Expected Count}}{\sqrt{\text{Variance}}} ) These residuals are independent of sequencing depth and are used for downstream analysis.
Protocol II: Feature Selection using Deviance
This protocol uses a deviance-based method to select highly informative genes directly from raw counts, minimizing biases introduced by transformation [24].
Detailed Methodology:
- Input Data: Use the raw count matrix without prior normalization.
- Deviance Calculation: For each gene, calculate the binomial deviance. This metric quantifies how poorly the gene's expression profile fits a constant (null) model across all cells. Genes with high deviance are highly variable and informative.
- Gene Ranking: Rank all genes in descending order of their binomial deviance value.
- Feature Selection: Select the top (n) genes (e.g., 2,000-4,000) from the ranked list for all subsequent dimensionality reduction and analysis.
The following workflow diagram illustrates the logical sequence of preprocessing steps, from raw data to a matrix ready for dimensionality reduction.
Workflow for scRNA-seq Preprocessing
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions and Computational Tools
| Item | Function | Example Use |
|---|---|---|
| Reference Transcriptome | A pre-assembled collection of genomic sequences and annotations for a species. | Used during primary analysis (e.g., by Cell Ranger) to align sequencing reads and identify transcripts [21]. |
| Spike-in RNA Controls | Exogenous RNA molecules added in known quantities to the cell lysate. | Serves as a standard baseline for technical variation estimation and normalization, e.g., in BASiCS [18] [22]. |
| UMIs (Unique Molecular Identifiers) | Short random nucleotide sequences that label individual mRNA molecules. | Corrects for PCR amplification biases during library preparation, enabling accurate digital counting of transcripts [18] [21]. |
| Cell Barcodes | Short nucleotide sequences that uniquely label each cell. | Allows sequencing reads from a single cell to be pooled and later computationally demultiplexed [21]. |
| Scanpy / Seurat | Comprehensive software toolkits for single-cell data analysis. | Provide integrated functions for the entire analysis workflow, including normalization, feature selection, dimensionality reduction, and clustering [19] [24] [22]. |
Implementation and Downstream Integration
The choice of normalization and feature selection methods directly influences the quality of the low-dimensional embedding. For instance, it has been shown that Highly Variable Gene selection is effective for producing high-quality data integrations and query mappings [23]. Furthermore, novel model-based dimensionality reduction techniques like scGBM, which directly models raw counts using a Poisson bilinear model, are emerging as powerful alternatives to the standard workflow of transformation followed by PCA, as they can better capture biological signal and avoid artifacts [11].
The following diagram summarizes the integration of these preprocessing steps into the broader scRNA-seq analysis pipeline, leading to biological insights.
Full scRNA-seq Analysis Pipeline
There is no single best-performing method for all datasets and biological questions. Therefore, it is considered good practice to test different normalization and feature selection strategies, using metrics from downstream analyses—such as cluster separation, conservation of biological variation, and batch effect removal—to guide the selection of the most appropriate method for a given study [23] [18] [22].
Unsupervised Clustering and Cell Type Annotation Strategies
Single-cell RNA sequencing (scRNA-seq) has revolutionized the study of cellular heterogeneity by enabling the transcriptomic profiling of individual cells [25]. A foundational application of this technology is the identification of distinct cell types and states through unsupervised clustering, a process that groups cells based on transcriptional similarity without prior biological knowledge [26]. Following clustering, the critical step of annotation assigns biological identity to these computationally derived groups, bridging the gap between statistical patterns and cellular function [8] [26].
This protocol details a standardized workflow for unsupervised clustering and cell type annotation, framed within the broader context of scRNA-seq data analysis research. It is designed for researchers, scientists, and drug development professionals seeking to delineate cellular populations in complex tissues, such as those found in tumor microenvironments, stem cell niches, and developing organs [27]. The guidance integrates established best practices with emerging methodologies, including the novel Average Overlap metric for refining cluster annotation [26].
Experimental Design and Prerequisites
Key Considerations Before Starting
Successful single-cell analysis requires careful pre-planning. Two principal requirements must be met prior to embarking on a project:
- Genomic Resource Availability: The sequencing data can only be interpreted if sequences can be assigned to gene models. Mapping reads to a genome with complete gene annotations is ideal. If unavailable, a high-quality transcriptome assembly must be generated [28].
- Cell Suspension Protocol: Generating quality single-cell or single-nuclei suspensions from the tissue of interest is a non-trivial hurdle and may require months of wet-lab optimization. The decision to sequence single cells or single nuclei depends on the biological question. Single cells generally capture more mRNA, providing a broader view of the transcriptome, while single nuclei are better for difficult-to-dissociate tissues (e.g., neurons) and allow for multiome studies combining transcriptomics with ATAC-seq [28].
Experimental Design for Valid Batch Effect Correction
Batch effects are a major challenge in scRNA-seq. To ensure biological variability can be separated from technical artifacts, the experimental design must allow for batch effect correction. The BUSseq method mathematically proves that true biological variance can be separated under three valid experimental designs [29]:
- Completely Randomized Design: Each batch measures all cell types. This is the most robust design but can be costly or impractical.
- Reference Panel Design: A reference batch contains all cell types, while other batches may contain subsets.
- Chain-Type Design: Batches are linked by shared cell types in a chain-like manner, without requiring a single universal reference.
A completely confounded design, where batch and cell type are perfectly correlated, is non-identifiable, and batch effects cannot be corrected through any computational method [29].
Wet-Lab Protocols and Reagent Solutions
The primary steps in scRNA-seq encompass single-cell isolation, cell lysis, reverse transcription, cDNA amplification, and library preparation [27]. Commercial kits and reagents now exist for all wet-lab steps, making this technology accessible to non-specialists [25].
Sample Preparation and Cell Isolation
The initial stage involves extracting viable, individual cells from the tissue of interest. For tissues where dissociation is challenging, or when working with frozen samples, single-nuclei RNA-seq (snRNA-seq) is a viable alternative. "Split-pooling" techniques using combinatorial indexing can process up to millions of cells without expensive microfluidic devices [27] [25].
Molecular Barcoding and Amplification
After RNA is converted to cDNA, the molecules are amplified by polymerase chain reaction (PCR) or in vitro transcription (IVT). To mitigate amplification biases, Unique Molecular Identifiers (UMIs) are used to label each individual mRNA molecule during reverse transcription, improving the quantitative accuracy of the data [27]. Protocols like CEL-Seq, MARS-Seq, Drop-Seq, inDrop-Seq, and 10x Genomics have incorporated UMIs [27].
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Commercially Available Single-Cell Solutions
| Commercial Solution | Capture Platform | Throughput (Cells/Run) | Key Features and Applications |
|---|---|---|---|
| 10x Genomics Chromium | Microfluidic oil partitioning | 500 – 20,000 [28] | High capture efficiency (70-95%); supports cells, nuclei, and fixed material [28]. |
| BD Rhapsody | Microwell partitioning | 100 – 20,000 [28] | Allows for larger cell sizes (<100 µm); enables targeted transcript detection [28]. |
| Parse Evercode | Multiwell-plate | 1,000 – 1M [28] | Very low cost per cell; ideal for large-scale projects; requires high cell input [28]. |
| Fluent/PIPseq (Illumina) | Vortex-based oil partitioning | 1,000 – 1M [28] | No microfluidics hardware needed; no restrictions related to cell size [28]. |
Computational Analysis Workflow
The computational analysis of scRNA-seq data begins with a count matrix (cells x genes) and proceeds through a series of steps to identify and annotate cell types.
Diagram 1: scRNA-seq Analysis Workflow. This flowchart outlines the key computational steps from raw data to biological interpretation, highlighting critical transitions between major stages.
Pre-Processing and Quality Control
Before analysis, the count matrix must be examined to remove poor-quality cells, which add technical noise and obscure biological signals [30]. QC is typically performed based on three key metrics, visualized in the diagram below.
Diagram 2: Quality Control Metrics. This diagram illustrates the three primary QC covariates used to filter low-quality cells and the biological or technical phenomena they indicate.
- QC Metrics and Thresholding: The distributions of these QC covariates are examined for outliers, which are filtered out by thresholding [8]. As detailed in Diagram 2, cells with low counts/genes and high mitochondrial content often indicate dead or dying cells, while cells with very high counts/genes may be doublets (multiple cells captured as one) [8]. These metrics must be considered jointly, as interpreting any one in isolation can lead to misinterpretation [8]. Thresholds should be as permissive as possible to avoid filtering out viable cell populations unintentionally.
Data Normalization, Feature Selection, and Dimensionality Reduction
Following QC, the data is normalized to remove technical variations (e.g., in sequencing depth) and make expression levels comparable across cells [8]. This is often achieved by scaling counts to a constant total per cell (e.g., 10,000) and log-transforming the result.
- Feature Selection: To reduce noise and computational complexity, analysis focuses on Highly Variable Genes (HVGs) that drive cell-to-cell heterogeneity. These genes are more likely to contain biologically meaningful information for distinguishing cell types [8].
- Dimensionality Reduction: Principal Component Analysis (PCA) is applied to the HVGs to create a lower-dimensional representation that captures the major axes of variation in the data. These principal components are then used for unsupervised clustering and for further non-linear dimensionality reduction with methods like UMAP or t-SNE, which provide 2D or 3D visualizations of cell relationships [8].
Protocol for Unsupervised Clustering
Clustering groups cells based on their transcriptional similarity in the reduced dimensional space (e.g., the top principal components). A common and effective method is the graph-based approach.
Materials (Computational): A normalized and scaled single-cell object in R (Seurat) or Python (Scanpy).
Procedure:
- Construct a k-Nearest Neighbor (k-NN) Graph: Model the cellular data as a graph, where each cell is a node. Connect each cell to its
kmost similar cells (default k=20 is often a good starting point) based on Euclidean distance in PCA space. - Refine Edge Weights: Apply the Jaccard similarity or a similar method to refine the edge weights between cells, reflecting the shared overlap of their neighborhoods.
- Community Detection: Use a community detection algorithm, such as the Louvain or Leiden algorithm, to partition the k-NN graph into groups of highly interconnected cells. These groups are the resulting clusters.
- Resolution Parameter: The
resolutionparameter controls the granularity of the clustering. A lower resolution (e.g., 0.2-0.8) yields fewer, broader clusters, while a higher resolution (e.g., 0.8-1.5) yields more, finer clusters. This should be tuned based on the biology of the system.
Cluster Annotation and the Average Overlap Metric
After clustering, the final and most critical step is to assign biological identities to the clusters. This is typically done by identifying marker genes for each cluster—genes that are statistically over-expressed in one cluster compared to all others.
Table 2: Marker Gene Identification and Annotation Methods
| Method | Principle | Application Context |
|---|---|---|
| Differential Expression Testing | Statistical tests (e.g., Wilcoxon rank-sum test) to find genes enriched in each cluster. | Standard, first-pass annotation; works well for broad cell types. |
| Reference-Based Annotation | Compare cluster gene expression profiles to curated reference datasets (e.g., Celldex, SingleR). | Rapid, automated annotation; useful for well-characterized systems (e.g., human, mouse). |
| Average Overlap Metric (AOM) | Compares ranked lists of marker genes in a top-weighted manner to define distances between clusters [26]. | Refining annotation for highly similar populations; resolving subtle heterogeneity. |
Annotation Protocol:
- Find Marker Genes: For each cluster, perform differential expression analysis against all other cells to generate a ranked list of marker genes.
- Initial Annotation: Use the top marker genes to query cell type-specific databases (e.g., PanglaoDB, CellMarker) and published literature for known cell types.
- Refine with Average Overlap (AOM): A single clustering resolution may not perfectly capture all biological populations. The AOM provides a quantitative measure of cluster similarity based on their marker gene rankings [26]. Calculate the AOM between neighboring clusters. A high AOM suggests the clusters are highly similar and might represent a single, coherent cell type or a subtle substate. This can guide decisions on whether to merge clusters or re-cluster at a different resolution. This approach has been shown to enable "robust, reproducible characterization... in highly homogeneous populations," such as T-cell development stages [26].
The following diagram illustrates this iterative annotation process.
Diagram 3: Cluster Annotation and Refinement. This workflow outlines the process of annotating clusters based on marker genes and using the Average Overlap Metric (AOM) to decide whether to merge similar clusters for a final, biologically meaningful annotation.
Downstream Analysis and Advanced Applications
With annotated cell types, researchers can proceed to high-level biological interpretation. Key downstream analyses include:
- Differential Expression Analysis: Comparing gene expression between specific cell types across conditions (e.g., healthy vs. diseased).
- Trajectory Inference: Modeling dynamic processes like differentiation by ordering cells along a pseudotemporal path.
- Cell-Cell Communication: Predicting interactions between different cell types based on ligand-receptor expression.
These analyses, built upon a robust foundation of clustering and annotation, can uncover novel biology, identify therapeutic targets, and characterize disease mechanisms, ultimately advancing drug discovery and personalized medicine [27].
Advanced Analytical Techniques: Machine Learning and Applications in Drug Discovery
Leveraging Machine Learning for Dimensionality Reduction and Trajectory Inference
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the decoding of gene expression profiles at the level of individual cells, thereby revealing cellular heterogeneity and complex biological processes that are masked in bulk sequencing approaches [31] [32]. However, the high-dimensionality and inherent sparsity of scRNA-seq data—where each cell is represented by expressions of thousands of genes—present significant computational challenges for analysis and interpretation [33]. Machine learning (ML) has emerged as a core computational framework to address these challenges, providing powerful tools for extracting biologically meaningful insights from complex single-cell transcriptomics data [31].
Two of the most critical applications of ML in scRNA-seq analysis are dimensionality reduction and trajectory inference. Dimensionality reduction techniques transform high-dimensional gene expression data into lower-dimensional spaces, preserving essential biological information while facilitating visualization and downstream analysis [33]. Trajectory inference methods order cells along pseudotemporal trajectories to reconstruct dynamic biological processes such as development, differentiation, and disease progression from snapshot data [34]. The integration of these ML-driven approaches has become fundamental to unlocking the potential of scRNA-seq across diverse research domains, from fundamental biology to precision medicine and drug discovery [31] [35].
This application note provides a comprehensive overview of current ML methodologies for dimensionality reduction and trajectory inference in scRNA-seq analysis. We present structured comparisons of algorithmic performance, detailed experimental protocols for implementation, visualization of key analytical workflows, and a curated toolkit of research reagents and computational solutions. The content is framed within the broader context of advancing single-cell data analysis research, with particular emphasis on practical implementation for researchers, scientists, and drug development professionals.
Machine Learning for Dimensionality Reduction in scRNA-seq Data
Dimensionality reduction serves as an essential preprocessing step in scRNA-seq analysis pipelines, condensing thousands of gene dimensions into a manageable set of latent features that capture the primary sources of biological variation [33]. Both classical linear methods and advanced non-linear ML approaches have been adapted or developed specifically to address the statistical characteristics of single-cell data, including high dimensionality, sparsity, over-dispersion, and excessive zero counts (dropout events) [11].
Principal Component Analysis (PCA) represents the most widely used linear dimensionality reduction technique. PCA performs an orthogonal linear transformation of the original gene expression space to create new uncorrelated variables (principal components) that capture decreasing proportions of the total variance [33]. However, standard PCA applied to transformed count data can induce spurious heterogeneity and mask true biological variability [11]. Model-based alternatives such as GLM-PCA and scGBM (single-cell Poisson bilinear model) directly model count distributions to avoid transformation artifacts and better capture biological signal [11].
Non-linear dimensionality reduction methods have gained prominence for their ability to capture complex manifolds and biological relationships. t-Distributed Stochastic Neighbor Embedding (t-SNE) minimizes the Kullback-Leibler divergence between probability distributions in high and low dimensions, emphasizing the preservation of local neighborhood structures [31] [36]. Uniform Manifold Approximation and Projection (UMAP) applies cross-entropy loss to balance both local and global structure preservation, offering improved computational efficiency and global coherence compared to t-SNE [31] [36]. More recently, Pairwise Controlled Manifold Approximation (PaCMAP) and TRIMAP have incorporated additional distance-based constraints to enhance preservation of both local detail and long-range relationships [36].
Deep learning architectures, particularly autoencoders (AEs) and variational autoencoders (VAEs), provide highly flexible frameworks for non-linear dimensionality reduction. These neural network models learn to compress data through an encoder network to a low-dimensional latent space, then reconstruct the input through a decoder network [37] [35]. The boosting autoencoder (BAE) represents a recent innovation that combines the advantages of unsupervised deep learning with boosting for formalizing structural assumptions, enabling the identification of small gene sets that explain latent dimensions [37].
Table 1: Performance Comparison of Dimensionality Reduction Methods for scRNA-seq Data
| Method | Category | Key Algorithmic Features | Strengths | Limitations |
|---|---|---|---|---|
| PCA | Linear | Orthogonal linear transformation, variance maximization | Computationally efficient, interpretable components | May miss non-linear relationships, sensitive to data transformation |
| scGBM | Model-based | Poisson bilinear model, iteratively reweighted SVD | Directly models counts, quantifies uncertainty, scales to millions of cells | Complex implementation, longer runtime than PCA [11] |
| t-SNE | Non-linear | Kullback-Leibler divergence minimization, focus on local structure | Excellent visualization of local clusters, captures fine-grained patterns | Computational intensity, loss of global structure, stochastic results [36] |
| UMAP | Non-linear | Cross-entropy optimization, Riemannian geometry | Preservation of global structure, faster than t-SNE | Parameter sensitivity, potential artifactual connections [36] |
| PaCMAP | Non-linear | Pairwise distance preservation with three neighborhood types | Balanced local/global preservation, robust performance | Less established in biological domains [36] |
| Autoencoder | Deep Learning | Neural network encoder-decoder architecture, reconstruction loss | Flexibility to capture complex patterns, customizable architectures | Black box nature, computational demands, requires large data [37] |
| BAE | Deep Learning | Componentwise boosting encoder, structural constraints | Identifies sparse gene sets, incorporates biological assumptions | Complex implementation, specialized use cases [37] |
Recent benchmarking studies evaluating dimensionality reduction methods on drug-induced transcriptomic data from the Connectivity Map (CMap) dataset have provided empirical performance comparisons across multiple experimental conditions [36]. The study evaluated 30 different DR methods using internal cluster validation metrics (Davies-Bouldin Index, Silhouette score, Variance Ratio Criterion) and external validation metrics (Normalized Mutual Information, Adjusted Rand Index) to assess their ability to preserve biological structures.
Table 2: Benchmarking Performance of Top Dimensionality Reduction Methods on Drug Response Data [36]
| Method | Internal Validation (DBI) | Internal Validation (Silhouette) | External Validation (NMI) | Dose-Response Sensitivity | Computational Efficiency |
|---|---|---|---|---|---|
| t-SNE | High | High | High | Strong | Moderate |
| UMAP | High | High | High | Moderate | High |
| PaCMAP | High | High | High | Moderate | High |
| TRIMAP | High | High | High | Low | High |
| PHATE | Moderate | Moderate | Moderate | Strong | Low |
| Spectral | Moderate | Moderate | Moderate | Strong | Low |
| PCA | Low | Low | Low | Low | High |
The benchmarking results demonstrated that PaCMAP, TRIMAP, t-SNE, and UMAP consistently ranked among the top performers across multiple datasets and evaluation metrics, particularly excelling in separating distinct cell types and grouping drugs with similar molecular targets [36]. However, most methods struggled with detecting subtle dose-dependent transcriptomic changes, where Spectral, PHATE, and t-SNE showed relatively stronger performance [36]. These findings highlight the importance of method selection based on specific analytical goals and data characteristics.
Experimental Protocol: Dimensionality Reduction with scGBM
Principle: The scGBM (single-cell generalized bilinear model) approach performs dimensionality reduction by directly modeling UMI count data using a Poisson bilinear model, avoiding transformation-induced artifacts and providing uncertainty quantification for downstream analyses [11].
Materials and Reagents:
- Computational Environment: R (version 4.1.0 or higher) or Python (version 3.8 or higher)
- Required Packages: scGBM R package (https://github.com/phillipnicol/scGBM) or equivalent implementation
- Hardware Recommendations: Minimum 8GB RAM for datasets <10,000 cells; 16GB+ RAM for larger datasets
- Input Data: Raw UMI count matrix (cells × genes) in sparse matrix format
Procedure:
- Data Preparation:
- Load the UMI count matrix, ensuring genes are in rows and cells in columns
- Filter low-quality cells based on mitochondrial percentage, total UMI counts, and detected gene counts
- Remove genes expressed in fewer than 10 cells to reduce noise
Model Initialization:
Parameter Estimation:
- Execute the iteratively reweighted singular value decomposition algorithm
- Monitor convergence via the Poisson deviance (typically requires 20-50 iterations)
- Extract the low-dimensional embedding (factor scores) and factor loadings
Uncertainty Quantification:
- Compute posterior uncertainties for each cell's latent position
- Calculate Cluster Cohesion Index (CCI) to assess confidence in cluster assignments
Downstream Analysis:
- Use the latent factors as input for clustering algorithms or visualization methods
- Identify genes associated with each factor through the loading matrix
Troubleshooting Tips:
- For convergence issues, reduce learning rate or increase number of iterations
- For memory limitations with large datasets, utilize the projection-based approximation
- Interpret factors by examining high-weight genes in the loading matrix
Experimental Protocol: Boosting Autoencoder for Interpretable Dimensionality Reduction
Principle: The Boosting Autoencoder (BAE) combines deep learning-based dimensionality reduction with componentwise boosting to incorporate structural assumptions and identify sparse sets of genes that characterize each latent dimension [37].
Materials and Reagents:
- Computational Environment: Python 3.7+ with PyTorch or TensorFlow
- Implementation: BAE code (https://github.com/NiklasBrunn/BoostingAutoencoder)
- Input Data: Normalized gene expression matrix (cells × genes)
Procedure:
- Data Preprocessing:
- Normalize gene expression counts using standard scRNA-seq preprocessing
- Select highly variable genes (typically 2,000-5,000 genes) to reduce computational burden
Model Configuration:
- Define encoder architecture as a linear transformation with boosting constraints
- Design decoder as a neural network with multiple hidden layers
- Specify structural constraints (disentanglement or temporal coupling) based on experimental design
Model Training:
Interpretation and Analysis:
- Extract sparse weight matrix from the boosting encoder
- Identify genes associated with each latent dimension through non-zero weights
- Project cells into latent space for visualization and clustering
Validation:
- Compare clustering results with known cell type markers
- Assess biological relevance of identified gene sets through enrichment analysis
Applications:
- Disentanglement Constraint: Identification of distinct cell types and corresponding marker genes
- Temporal Coupling: Analysis of developmental processes with time-series data
Machine Learning for Trajectory Inference in scRNA-seq Data
Trajectory inference (TI) methods aim to reconstruct dynamic biological processes by ordering cells along pseudotemporal trajectories based on gene expression similarity, enabling the study of cellular differentiation, development, and disease progression from snapshot scRNA-seq data [34]. While early TI methods treated pseudotime as a descriptive concept based on expression distance metrics, recent advances have focused on developing more principled model-based approaches with biophysical interpretations [34].
The evolution of TI methodologies has progressed from graph-based approaches that construct minimum spanning trees or principal graphs through expression space, to RNA velocity-based methods that leverage unspliced/spliced mRNA ratios to predict future cell states, and more recently to process time models that infer latent variables corresponding to the timing of cells subject to specific biophysical processes [34].
The Chronocell model represents a significant advancement in this evolution by formulating trajectories through a biophysical framework of cell state transitions [34]. Unlike descriptive pseudotime, Chronocell infers "process time" as a latent variable with intrinsic physical meaning relative to a specific cellular process. The model is identifiable, making parameter inference meaningful, and can interpolate between trajectory inference (for continuous cell states) and clustering (for discrete states) based on data characteristics [34].
Key innovations in modern TI methods include:
- Biophysical Meaning: Parameters such as degradation rates and transition probabilities have direct biological interpretations
- Model Identifiability: Ensuring that inferred parameters correspond to unique solutions
- Model Assessment: Quantitative frameworks for determining whether data support continuous trajectory or discrete cluster models
- Integration with RNA Velocity: Combining snapshot data with kinetic models of RNA splicing
Table 3: Comparison of Trajectory Inference Methodologies
| Method Category | Representative Algorithms | Underlying Principle | Interpretation of Time | Key Assumptions |
|---|---|---|---|---|
| Graph-Based | Monocle, Slingshot | Minimum spanning trees through expression space | Descriptive pseudotime based on expression distance | Continuous biological process exists in data |
| RNA Velocity | scVelo, Velocyto | Kinetic modeling of unspliced/spliced mRNA ratios | Directional flow based on RNA metabolism | Splicing kinetics are consistent across cells |
| Process Models | Chronocell, VeloCycle | Biophysical models of cell state transitions | Process time with physical interpretation | Cells share common dynamic process |
Experimental Protocol: Trajectory Inference with Chronocell
Principle: Chronocell implements a process time model that formulates trajectories through biophysical modeling of cell state transitions, inferring latent variables corresponding to the timing of cells subject to a specific cellular process [34].
Materials and Reagents:
- Computational Environment: MATLAB or Python with specialized Chronocell implementation
- Software Availability: https://github.com/pachterlab/FGP_2024
- Input Data: Processed scRNA-seq count matrix with preliminary cell type annotations
Procedure:
- Data Preprocessing:
- Perform standard scRNA-seq preprocessing (normalization, highly variable gene selection)
- Compute preliminary dimensionality reduction (PCA) and clustering
- Annotate broad cell states based on marker genes
Model Initialization:
- Specify the number of cell states and possible transitions
- Initialize process time parameters based on prior knowledge or heuristic ordering
Parameter Estimation:
Model Selection and Assessment:
- Compare trajectory model against cluster models using likelihood ratio tests
- Assess model fit through residual analysis and posterior predictive checks
- Validate process time ordering using known marker gene dynamics
Interpretation and Visualization:
- Extract process time estimates for each cell
- Plot gene expression dynamics along process time
- Identify transition points between cell states
Validation and Troubleshooting:
- Ground Truth Validation: Compare with time-series data where available
- Sensitivity Analysis: Assess robustness to parameter initialization
- Model Adequacy Checking: Verify that continuous trajectory model is more appropriate than discrete clusters for the data
- Circularity Avoidance: Ensure that the same data is not used for both trajectory inference and differential expression testing without proper cross-validation
Specialized Protocol: Trajectory Inference with Compositional Data Analysis
Principle: Compositional Data Analysis (CoDA) provides an alternative statistical framework for trajectory inference by treating scRNA-seq data as compositions of log-ratios between components, which can improve robustness to dropout events and other technical artifacts [38].
Materials and Reagents:
- R Package: CoDAhd (https://github.com/GO3295/CoDAhd)
- Input Data: Raw UMI count matrix
Procedure:
- Count Addition:
- Apply specific count addition schemes (e.g., SGM) to handle zero values
- Convert raw counts to compositions
Log-Ratio Transformation:
- Compute centered log-ratio (CLR) transformation
- Alternative: Use other log-ratio transformations (ALR, ILR) based on data characteristics
Trajectory Analysis:
- Apply trajectory inference algorithms (e.g., Slingshot) to CLR-transformed data
- Compare results with conventional normalization approaches
Applications: Particularly valuable for datasets with high dropout rates or when conventional methods produce biologically implausible trajectories.
Integrated Workflow and Visualization
The analytical workflow for scRNA-seq analysis typically follows a sequential pipeline where dimensionality reduction precedes trajectory inference, with each step informing subsequent analyses. The following diagram illustrates this integrated workflow and the key decision points:
Diagram 1: Integrated Workflow for Dimensionality Reduction and Trajectory Inference. The diagram illustrates the sequential analytical steps in scRNA-seq analysis, with key decision points for method selection based on data characteristics and research objectives.
The relationship between different dimensionality reduction methods and their performance characteristics can be visualized through the following comparative framework:
Diagram 2: Method Characteristics Across Dimensionality Reduction Approaches. The diagram compares key performance characteristics across major categories of dimensionality reduction methods, highlighting trade-offs and complementary strengths.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 4: Essential Computational Tools for Dimensionality Reduction and Trajectory Inference
| Tool Name | Category | Primary Function | Implementation | Key Features |
|---|---|---|---|---|
| scGBM | Dimensionality Reduction | Model-based dimensionality reduction | R package | Direct count modeling, uncertainty quantification, scalability to millions of cells [11] |
| Boosting Autoencoder (BAE) | Dimensionality Reduction | Interpretable deep learning dimensionality reduction | Python/PyTorch | Sparse gene sets, structural constraints, disentangled dimensions [37] |
| Chronocell | Trajectory Inference | Process time modeling | MATLAB/Python | Biophysical interpretation, model identifiability, trajectory/clustering interpolation [34] |
| CoDAhd | Data Transformation | Compositional data analysis for scRNA-seq | R package | Centered log-ratio transformation, dropout robustness, improved trajectory inference [38] |
| scVelo | Trajectory Inference | RNA velocity analysis | Python | Kinetic modeling, dynamical inference, gene-shared time [34] |
| Slingshot | Trajectory Inference | Graph-based trajectory inference | R package | Minimum spanning trees, simultaneous lineage identification [38] |
Table 5: Experimental Design Considerations for Method Selection
| Research Scenario | Recommended Dimensionality Reduction | Recommended Trajectory Inference | Rationale |
|---|---|---|---|
| Standard cell type identification | PCA or scGBM | Graph-based (Slingshot) | Computational efficiency, established benchmarks |
| Rare cell population detection | scGBM or BAE | Process time (Chronocell) | Enhanced sensitivity to small cell groups, uncertainty quantification |
| High dropout rate datasets | scGBM or CoDA-transformed PCA | CoDA-enhanced trajectory inference | Robustness to technical zeros, compositionally aware |
| Biophysical parameter estimation | Model-based (scGBM) | Process time (Chronocell) | Parameter interpretability, kinetic modeling |
| Developmental time series | BAE with temporal constraints | RNA velocity or process time | Temporal structure incorporation, directional information |
| Large-scale datasets (>1M cells) | scGBM or UMAP | Graph-based methods | Computational scalability, efficient neighbor detection |
Machine learning approaches for dimensionality reduction and trajectory inference have fundamentally transformed the analysis of single-cell RNA sequencing data, enabling researchers to extract profound biological insights from increasingly complex and large-scale datasets. The field has evolved from purely descriptive visualizations and orderings to principled model-based approaches that incorporate biophysical meaning and uncertainty quantification.
The current landscape offers a diverse toolkit of methods, each with distinct strengths and optimal application domains. For dimensionality reduction, researchers can select from computationally efficient linear methods (PCA), count-aware model-based approaches (scGBM), visualization-optimized non-linear techniques (UMAP, t-SNE), or interpretable deep learning architectures (BAE) based on their specific analytical needs. Similarly, trajectory inference has progressed from graph-based pseudotime orderings to biophysically grounded process time models (Chronocell) that provide meaningful parameter estimates and rigorous model assessment.
Future directions in this rapidly advancing field will likely focus on several key areas: (1) enhanced integration of multi-omics data types within unified dimensionality reduction frameworks; (2) development of increasingly interpretable and biologically constrained models; (3) improved scalability to accommodate the growing size of single-cell datasets; and (4) tighter coupling between experimental design and computational analysis to ensure biological validity. As these methodologies continue to mature, they will further empower researchers to unravel the complexities of cellular systems, accelerating discoveries in basic biology, disease mechanisms, and therapeutic development.
By providing structured comparisons, detailed protocols, and practical implementation guidelines, this application note serves as a comprehensive resource for researchers navigating the evolving landscape of machine learning approaches for single-cell RNA sequencing analysis. The integration of these computational methodologies with experimental single-cell technologies will continue to drive innovations in precision medicine and therapeutic development.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biomedical research by enabling the investigation of transcriptional profiles at the individual cell level, moving beyond the limitations of bulk RNA sequencing which masks cellular heterogeneity [39]. Since its inception in 2009, scRNA-seq has evolved into a powerful tool that captures the diversity within tissues, organs, and individuals, generating millions to billions of datapoints per experiment [40] [39]. In the high-stakes field of drug discovery, where development takes approximately 10-15 years and costs between $900 million to over $2 billion per drug, scRNA-seq offers unprecedented resolution to dissect cellular mechanisms and streamline development [40]. This technology provides nuanced insights into drug targets, biomarkers, and patient responses, potentially reducing the staggering attrition rates in clinical trials by identifying pharmacokinetic and toxicity issues earlier in the process [40]. This Application Note details standardized protocols and applications for integrating scRNA-seq throughout the drug discovery pipeline, from initial target identification to clinical biomarker development, providing researchers with practical methodologies to leverage this transformative technology.
scRNA-seq in Target Identification and Validation
Target identification and validation represent the foundational stage of drug discovery, and scRNA-seq significantly enhances this process by revealing cell-type-specific gene expression patterns in disease-relevant tissues. A 2024 retrospective analysis from the Wellcome Institute demonstrated that drug targets with cell-type-specific expression in disease-relevant tissues are robust predictors of clinical trial progression from Phase I to Phase II [40]. By analyzing 30 diseases and 13 tissues, researchers established scRNA-seq as a predictive tool for prioritizing targets with higher success potential.
Experimental Protocol: Target Discovery Using scRNA-seq
Sample Preparation and Single-Cell Isolation
- Tissue Dissociation: Mechanically and enzymatically dissociate fresh tissue samples (e.g., tumor biopsies) to create single-cell suspensions. For challenging samples or frozen tissues, single-nuclei RNA sequencing (snRNA-seq) provides a viable alternative [39].
- Cell Viability Assessment: Assess viability using trypan blue staining, aiming for >90% viability.
- Cell Capture: Utilize droplet-based systems (e.g., 10x Genomics Chromium) for high-throughput capture or plate-based fluorescence-activated cell sorting (FACS) for larger cells (>30μm) [39]. For massive-scale experiments, combinatorial indexing platforms (e.g., Parse Biosciences Evercode) can barcode up to 10 million cells across thousands of samples [40].
- Quality Control: Retain cells with nFeature_RNA > 200 and < 5000, and mitochondrial gene percentage (percent.mt) < 5% to exclude low-quality cells and potential doublets [41].
Library Preparation and Sequencing
- cDNA Synthesis and Amplification: Perform reverse transcription with template-switching oligos for PCR-based amplification or employ in vitro transcription (IVT) for linear amplification [27].
- Molecular Barcoding: Incorporate Unique Molecular Identifiers (UMIs) during reverse transcription to correct for PCR amplification biases and enable accurate transcript quantification [27].
- Library Construction: Prepare sequencing libraries using 3' end-enriched protocols for cost-effective expression profiling or full-length transcripts for isoform-level analysis [39].
- Sequencing: Sequence on high-throughput platforms (Illumina NovaSeq) with sufficient depth (e.g., 50,000 reads/cell) to capture rare cell types and low-abundance transcripts.
Computational Analysis for Target Identification
- Data Preprocessing: Filter low-quality cells, normalize using SCTransform, and detect highly variable genes (HVGs) [41].
- Dimensionality Reduction and Clustering: Perform principal component analysis (PCA), followed by graph-based clustering (Louvain algorithm) and visualization with UMAP/t-SNE [42] [41].
- Differential Expression Analysis: Identify cluster-specific markers using Wilcoxon rank-sum test with |log2FC| > 1 and adjusted p-value < 0.01 [41].
- Target Prioritization: Prioritize targets exhibiting specific expression in disease-relevant cell types, involvement in key pathways, and association with clinical outcomes.
Integrated CRISPR-scRNA-seq for Target Validation
For target validation, combine scRNA-seq with CRISPR screening to map regulatory elements and gene functions:
- CRISPR Perturbation: Conduct pooled CRISPR screens targeting candidate genes in disease models.
- Single-Cell Profiling: Sequence individual perturbed cells to assess transcriptomic consequences.
- Data Integration: Analyze how perturbations affect gene expression networks and pathways, providing functional validation of therapeutic targets [40]. This approach has been successfully applied to profile approximately 250,000 primary CD4+ T cells, systematically mapping regulatory element-to-gene interactions [40].
Table 1: Key Reagents for Target Identification and Validation
| Research Reagent | Function | Example Products |
|---|---|---|
| Tissue Dissociation Kits | Generate single-cell suspensions | Miltenyi Biotec GentleMACS |
| Cell Capture Reagents | Isolate individual cells | 10x Genomics Chromium Next GEM |
| Reverse Transcription Master Mix | Convert RNA to cDNA | Parse Biosciences Evercode |
| Library Preparation Kits | Prepare sequencing libraries | Smart-Seq2, CEL-Seq2 |
| CRISPR Screening Libraries | Introduce genetic perturbations | Brunello CRISPRko library |
scRNA-seq in Drug Screening and Mechanism of Action Studies
Traditional drug screening relying on general readouts like cell viability lacks comprehensive detail on cellular responses. scRNA-seq enables detailed cell-type-specific gene expression profiling, essential for understanding drug mechanisms and identifying subtle efficacy and resistance patterns [40]. High-throughput screening now incorporates scRNA-seq for multi-dose, multiple experimental conditions, and perturbation analyses, providing richer data that support comprehensive insights into cellular responses and pathway dynamics [40] [43].
Experimental Protocol: High-Throughput Drug Screening with scRNA-seq
Study Design
- Cell Models: Use patient-derived organoids or primary cells to maintain physiological relevance. For cancer studies, include multiple cell lines representing disease heterogeneity [44].
- Compound Library: Screen compound libraries across multiple concentrations and time points.
- Experimental Conditions: Include appropriate controls (DMSO vehicle, positive controls) and replication (minimum n=3).
- Multiplexing: Incorporate sample barcoding (e.g., MULTI-seq) to process multiple conditions simultaneously, reducing batch effects and costs.
Drug Perturbation and Processing
- Treatment Protocol: Treat cells with compounds across a concentration range (e.g., 1nM-10μM) for 24-72 hours.
- Cell Harvesting: Trypsinize adherent cells or collect suspension cells at endpoint.
- Cell Staining: For multimodal data, include antibody staining (CITE-seq) for surface protein expression.
- Pooling and Sequencing: Pool barcoded samples before library preparation and sequence at appropriate depth.
Computational Analysis for Drug Screening
- Data Integration: Harmonize data across conditions using integration methods (Harmony, Seurat CCA) [41].
- Differential Expression Analysis: Identify compound-induced gene expression changes using pseudobulk methods (DESeq2, edgeR) or mixed models.
- Pathway Analysis: Perform Gene Set Enrichment Analysis (GSEA) to identify affected biological pathways.
- Cell State Transitions: Construct pseudotime trajectories (Monocle, Slingshot) to model how compounds influence cellular differentiation states [42] [41].
Case Study: Large-Scale Cytokine Screening
A pioneering study demonstrated the power of scRNA-seq in large-scale perturbation screening, measuring 90 cytokine perturbations across 18 immune cell types from twelve donors, resulting in nearly 20,000 observed perturbations [40]. This generated a 10 million cell dataset with 1,092 samples in a single run, revealing that large sample sizes are essential to detect behavior of all cells, including rare types. When the authors downsampled a small PBMC subset (CD16 monocytes, only 5-10% of monocytes), cytokine effects were barely detectable in just 78 cells, but increasing the sample size to 2,500 significantly boosted detection of differentially expressed genes [40].
Table 2: Quantitative Results from Cytokine Perturbation Study
| Cell Population | Sample Size | DEGs Detected | Key Findings |
|---|---|---|---|
| CD16+ Monocytes | 78 cells | 2-5 DEGs | Limited detection power |
| CD16+ Monocytes | 2,500 cells | 50+ DEGs | Significant detection improvement |
| CD4+ Memory T cells | 3,000 cells | 45+ DEGs | Shared and unique response patterns |
| Rare cell types (<1%) | >1,000 cells | 15-30 DEGs | Requires sufficient cell numbers |
scRNA-seq for Biomarker Discovery and Patient Stratification
Biomarkers are objectively measurable characteristics of biological processes, with applications in prognosis, diagnosis, prediction, and treatment monitoring. While bulk transcriptomics has historically been used for biomarker discovery, it fails to capture cellular population complexity [40]. scRNA-seq advances this field by defining more accurate biomarkers through resolution of cellular heterogeneity, enabling more precise patient stratification and tailored therapeutic strategies [40] [45].
Experimental Protocol: Biomarker Identification from Clinical Samples
Cohort Selection and Sample Processing
- Patient Cohort: Prospectively enroll patients representing disease diversity, collecting relevant clinical metadata.
- Sample Collection: Process fresh tissues immediately or preserve by snap-freezing for snRNA-seq.
- Sample Processing: Process all samples using standardized protocols to minimize technical variability.
- Quality Assessment: Rigorously assess RNA quality (RIN >7) and cell viability before sequencing.
Cell Type-Specific Biomarker Identification
- Cell Atlas Construction: Generate a comprehensive cell atlas by clustering and annotating cell types using reference datasets (Blueprint, HPCA) [42].
- Differential Expression: Identify cell-type-specific markers differentially expressed between disease states or treatment responses.
- Survival Association: Correlate expression of candidate biomarkers with clinical outcomes using survival analysis (Cox regression) [41].
- Validation: Confirm findings using orthogonal methods (RT-qPCR, immunohistochemistry) in independent cohorts.
Case Study: Biomarker Heterogeneity in Breast Cancer Resistance
A 2025 study investigated biomarker heterogeneity linked to CDK4/6 inhibitor resistance in breast cancer using scRNA-seq of seven palbociclib-naïve luminal breast cancer cell lines and their resistant derivatives [44]. Researchers analyzed 10,557 cells with at least 2,000 genes expressed per cell, revealing that established resistance biomarkers (CCNE1, RB1, CDK6, FAT1, FGFR1) showed marked intra- and inter-cell-line heterogeneity. Transcriptional features of resistance were already observable in naïve cells, correlating with sensitivity levels (IC50) to palbociclib [44]. Resistant derivatives showed transcriptional clusters that significantly varied for proliferative, estrogen response signatures, or MYC targets, explaining why single biomarkers have failed clinically and highlighting the need for multi-gene signatures accounting for heterogeneity [44].
Data Analysis, Integration, and Visualization Pipeline
Computational Workflow for scRNA-seq Data
The analysis of scRNA-seq data requires specialized computational tools and workflows to transform raw sequencing data into biological insights. The following diagram illustrates the core analytical pipeline:
Table 3: Essential Computational Tools for scRNA-seq Analysis
| Analysis Step | Tool Options | Key Features |
|---|---|---|
| Quality Control | Seurat, Scanpy | Filtering by nFeature, nCount, percent.mt |
| Normalization | SCTransform, scran | Removal of technical variability |
| Dimensionality Reduction | PCA, UMAP, t-SNE | Visualization of high-dimensional data |
| Clustering | Louvain, Leiden | Identification of cell populations |
| Differential Expression | Wilcoxon test, MAST | Identification of marker genes |
| Trajectory Inference | Monocle, Slingshot | Reconstruction of differentiation paths |
| Cell-Cell Communication | CellChat, NicheNet | Inference of signaling networks |
| Data Integration | Harmony, Seurat CCA | Batch effect correction |
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 4: Key Research Reagent Solutions for scRNA-seq Studies
| Reagent Category | Specific Product Examples | Function in Workflow |
|---|---|---|
| Single-Cell Isolation Platforms | 10x Genomics Chromium, Parse Biosciences Evercode, BD Rhapsody | Partitioning cells/nuclei into reactions with barcodes |
| Library Preparation Kits | Smart-Seq2, CEL-Seq2, MARS-Seq | cDNA amplification and library construction |
| Enzymes & Master Mixes | Maxima H Minus Reverse Transcriptase, Template Switching Enzyme | Reverse transcription and cDNA amplification |
| Barcodes & Primers | Cell Multiplexing Oligos (CMO), Sample Multiplexing Oligos | Sample multiplexing and sample tracking |
| Quality Control Assays | Bioanalyzer, Fragment Analyzer, QuBit | Assessment of nucleic acid quality and quantity |
| Cell Viability Assays | Trypan Blue, Fluorescent Viability Dyes | Assessment of cell integrity pre-encapsulation |
| Reference Databases | Human Protein Atlas, Blueprint/ENCODE, HPCA | Cell type annotation and biological context |
This Application Note demonstrates the transformative power of scRNA-seq technology throughout the drug discovery pipeline, from initial target identification to clinical biomarker development. The protocols and case studies presented provide researchers with practical frameworks for implementing these approaches in their own work. As sequencing technologies continue to advance, with increasing throughput and decreasing costs, and as computational methods become more sophisticated through AI and machine learning integration, scRNA-seq is poised to become an indispensable tool in precision medicine, enabling the development of more effective, targeted therapies with higher clinical success rates [40] [39] [43]. The future of drug discovery lies in embracing cellular heterogeneity rather than averaging it away, and scRNA-seq provides the essential toolset for this paradigm shift.
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity and complex tissue ecosystems by enabling the profiling of gene expression at individual cell resolution [46]. In cancer research, this technology has proven particularly transformative for dissecting the tumor microenvironment (TME), a dynamic ecosystem composed of malignant cells and non-malignant components including immune infiltrates and stromal elements [47] [48]. The incredible sensitivity and specificity achieved by quantifying molecular alterations at single-cell resolution have led to unprecedented opportunities for uncovering the molecular mechanisms underlying disease pathogenesis and progression [49].
The transition from bulk RNA sequencing to scRNA-seq has fundamentally changed the field of tumor biology by providing a strategy to demonstrate TME heterogeneity and intercellular communication at the single-cell level [50]. This technological advancement is crucial because traditional bulk analyses obscure the nuances between rare subpopulations and cellular states, masking crucial biological details associated with disease development and treatment response [48]. The application of scRNA-seq in biomedical studies has advanced our diagnostic and therapeutic strategies, particularly in the context of cancer immunotherapy and personalized treatment approaches [49] [51].
Key Single-Cell Technologies and Methodologies
scRNA-seq Workflow and Platform Selection
The typical scRNA-seq workflow encompasses five critical steps: single-cell isolation and capture, cell lysis, reverse transcription, cDNA amplification, and library preparation [46]. Currently, two primary methodologies dominate the field: plate-based and droplet-based approaches. Plate-based methods such as SMART-seq2 utilize fluorescence-activated cell sorting (FACS) to isolate individual cells into multi-well plates, enabling full-length transcript sequencing which is particularly advantageous for detecting rare cell types and low-abundance genes [48]. In contrast, droplet-based methods including 10x Genomics Chromium, Drop-seq, and inDrop use microfluidic partitioning to encapsulate single cells with barcoded beads, allowing high-throughput processing of thousands of cells simultaneously while typically capturing only the 3' or 5' ends of transcripts [46] [48].
Table 1: Comparison of Major scRNA-seq Platforms and Their Applications in TME Studies
| Platform | Methodology | Transcript Coverage | Throughput | Key Applications in TME |
|---|---|---|---|---|
| SMART-Seq2 [46] | Plate-based | Full-length | Low (hundreds of cells) | Detection of rare cell populations, isoform usage analysis |
| 10x Genomics Chromium [49] [48] | Droplet-based | 3' or 5' counting | High (thousands of cells) | Comprehensive TME cell atlas construction, large-scale studies |
| Drop-Seq [46] | Droplet-based | 3' end | High | Tumor heterogeneity studies, cost-effective large-scale projects |
| Fluidigm C1 [46] | Microfluidics-based | Full-length | Medium | Precise cell handling, sensitive detection of low-abundance transcripts |
| inDrop [46] | Droplet-based | 3' end | High | Large-scale tumor infiltration studies |
Emerging Spatial Transcriptomics Technologies
Recent advancements in spatial omics technologies have enabled the detection of numerous markers within their tissue context, addressing a critical limitation of conventional scRNA-seq which loses spatial information during cell dissociation [48]. Imaging-based approaches such as ChipCytometry, CyCIF, and MICs use iterative cycles of antibody staining, imaging, and stripping with customized multiplexed panels. Next-generation sequencing-based methods including MERFISH, SeqFISH, Slide-seqV2, and Visium HD enable quantitative detection of up to 10,000 transcripts. Integrated platforms such as PhenoCycler (CODEX), DBiT-seq, CosMx, and Xenium now allow simultaneous detection of both protein and RNA markers, providing comprehensive spatial mapping of the TME [48].
Diagram 1: Comprehensive scRNA-seq Experimental Workflow for TME Analysis. The process begins with tissue dissociation and quality control, proceeds through single-cell isolation using various methodologies, continues with library preparation and sequencing, and concludes with comprehensive computational analysis.
Computational Analysis Framework
Core Data Processing and Quality Control
The computational analysis of scRNA-seq data begins with raw data processing including sequencing read quality control, read mapping, cell demultiplexing, and cell-wise unique molecular identifier (UMI) count table generation [49]. Standardized pipelines such as Cell Ranger for 10x Genomics Chromium and CeleScope for Singleron's systems are commonly employed, though alternative tools including UMI-tools, scPipe, zUMIs, and kallisto bustools are also available [49]. Quality control focuses on identifying and removing damaged cells, dying cells, stressed cells, and doublets using three primary metrics: total UMI count (count depth), number of detected genes, and the fraction of mitochondrial-derived counts per cell barcode [49]. Cells with low numbers of detected genes and low count depth typically indicate damaged cells, while a high proportion of mitochondrial counts suggests dying cells. Conversely, excessively high detected genes and count depth often indicate doublets [49].
Advanced Analytical Approaches for TME Dissection
Following quality control, basic data analysis includes normalization, feature selection, dimensionality reduction, cell clustering, and cell type annotation. Unsupervised clustering algorithms group cells based on transcriptome similarity, followed by annotation using canonical marker genes [49] [48]. Advanced analytical techniques then enable deeper investigation of TME biology:
Trajectory Inference: Pseudotemporal ordering of cells along differentiation trajectories reveals dynamic processes such as T-cell exhaustion, macrophage polarization, and tumor evolution [49] [52].
Cell-Cell Communication (CCC) Analysis: Computational tools leverage ligand-receptor interaction databases to infer intercellular communication networks within the TME, highlighting immune evasion mechanisms [49] [48].
Gene Regulatory Network (GRN) Inference: Single-cell network biology approaches reconstruct regulatory interactions using Boolean models, ordinary differential equations, and information theory to identify key transcriptional drivers of cellular states [52].
Copy Number Variation (CNV) Analysis: Tools like inferCNV estimate copy number variations from scRNA-seq data to distinguish malignant from non-malignant cells and investigate intra-tumoral heterogeneity [50].
Table 2: Essential Computational Tools for scRNA-seq Analysis in TME Studies
| Analysis Type | Tool/Platform | Key Function | Application in TME Research |
|---|---|---|---|
| Data Processing | Cell Ranger [49] | Raw data processing, alignment | Generation of UMI count matrices from 10x Genomics data |
| Quality Control | Seurat [49] [50] | Cell filtering, normalization | Identification of low-quality cells, doublet removal |
| Clustering & Visualization | Scater [49] | Dimensionality reduction, clustering | Identification of distinct cell populations within TME |
| Trajectory Inference | Monocle2 [50] | Pseudotemporal ordering | Reconstruction of cell state transitions, differentiation pathways |
| Cell-Cell Communication | CellPhoneDB [48] | Ligand-receptor interaction analysis | Mapping tumor-immune-stromal communication networks |
| CNV Analysis | inferCNV [50] | Copy number variation inference | Discrimination of malignant vs. non-malignant cells |
| Regulatory Networks | PySCENIC [50] | Gene regulatory network reconstruction | Identification of key transcription factors in tumor cells |
Diagram 2: Cellular Architecture of the Tumor Microenvironment. The TME comprises three major compartments: malignant cells with their genomic heterogeneity, diverse immune cell populations with both anti-tumor and pro-tumor functions, and stromal components that provide structural support and modulate ecosystem function through complex intercellular communication.
Application Notes: Case Studies in Tumor Heterogeneity
Revealing Immune Evasion Mechanisms in Osteosarcoma
A comprehensive scRNA-seq study of osteosarcoma (OS) TME analyzed data from seven primary tumors, two recurrent lesions, and two lung metastases, integrated with bulk RNA-seq data from 85 patients [50]. Researchers identified a novel population of tumor-educated "betrayer" dendritic cells characterized by CD83+CCR7+LAMP3+ markers, which were nearly absent in normal peripheral blood mononuclear cells [50]. Pseudotemporal trajectory analysis using Monocle2 revealed that these mature regulatory DCs (mregDCs) originated from conventional type 1 DCs (cDC1) and upregulated coinhibitory molecules including CD274 (PD-L1), LAG3, LGALS9, SIRPA, TIGIT, and PDCD1LG2 along the differentiation path [50]. These mregDCs specifically expressed chemokines CCL17, CCL19, and CCL22, creating a gradient that recruited regulatory T cells (Tregs) into the TME. Spatial analysis confirmed the physical juxtaposition of mregDCs and Tregs, with Treg density significantly higher within 100μm of mregDCs [50]. This study demonstrates how scRNA-seq can reveal novel immune evasion mechanisms with direct therapeutic implications.
Discovering Tissue-Specific Neuroendocrine Signatures
A groundbreaking scRNA-seq study of small cell neuroendocrine cervical carcinoma, the first such study worldwide for this rare and aggressive tumor type, revealed unique gene regulatory networks distinct from neuroendocrine carcinomas of the lung, small intestine, and liver [53]. Analysis of gene expression regulatory networks identified the transcription factor TFF3 as a key driver upregulating ELF3 expression [53]. This tissue-specific signature highlights how scRNA-seq can uncover origin-specific pathogenic mechanisms in histologically similar tumors, with direct implications for developing targeted therapies appropriate for each cancer type.
Computational Drug Repurposing for Cancer Immunotherapy
scRNA-seq enables innovative computational approaches for drug discovery by identifying specific cellular targets within the TME. Tools such as scDrug and scDrugPrio leverage single-cell data to predict tumor cell-specific cytotoxicity and prioritize drugs that reverse gene signatures associated with immune checkpoint inhibitor (ICI) non-responsiveness across diverse TME cell types [51]. This approach is particularly valuable for identifying combination therapies that can overcome resistance to ICIs, which show variable response rates across cancer types [51]. By focusing on patient-specific cellular profiles, these computational drug repurposing strategies facilitate personalized treatment approaches that target individual TME composition and functional states.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for scRNA-seq TME Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Viability Dyes (e.g., Propidium Iodide) [49] | Identification of live/dead cells | Critical for excluding dead cells during cell sorting; essential for data quality |
| UMI Barcodes [46] [49] | Molecular tagging of individual transcripts | Enables accurate transcript quantification and reduction of amplification bias |
| Cell Hash Tag Antibodies [49] | Sample multiplexing | Allows pooling of multiple samples, reducing batch effects and costs |
| FACS Antibody Panels [46] [48] | Cell surface marker detection | Facilitates targeted cell sorting for specific TME populations |
| mRNA Capture Beads [46] | Poly-A RNA capture | Foundation of droplet-based systems; critical for mRNA recovery efficiency |
| Reverse Transcriptase [46] | cDNA synthesis | Key determinant of library complexity and sensitivity |
| Template Switching Oligos [46] | cDNA amplification | Enables full-length transcript capture in SMART-seq2 protocols |
| Library Preparation Kits [49] | Sequencing library construction | Platform-specific optimized reagents for final library preparation |
The application of scRNA-seq technology to dissect the tumor microenvironment and cellular heterogeneity has fundamentally advanced cancer biology and therapeutic development. By enabling the precise characterization of cellular composition, transcriptional states, regulatory networks, and intercellular communication, this powerful approach provides unprecedented insights into tumor ecology. The integration of computational biology with sophisticated experimental methodologies continues to reveal novel therapeutic targets and resistance mechanisms, particularly in the context of immunotherapy. As spatial transcriptomics and multi-omics technologies mature, coupled with advanced computational tools for data integration and analysis, single-cell approaches will increasingly guide personalized treatment strategies and accelerate the development of more effective cancer therapies tailored to individual patient TME compositions.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biomedical science by enabling detailed exploration of gene expression at the cellular level, capturing inherent heterogeneity within samples [54]. However, cellular information extends well beyond the transcriptome, and a comprehensive understanding of cellular identity and function requires integration of multiple molecular modalities [55] [54]. Multi-omics integration, particularly combining scRNA-seq with DNA-based genomic information and Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq), provides a powerful approach to link genetic variation, epigenetic regulation, and transcriptional outcomes within the same biological system [56] [57]. This protocol details computational methodologies for integrating these modalities to uncover regulatory mechanisms, identify cell types, and understand disease pathogenesis.
Key Integration Methods and Applications
Table 1: Computational Methods for Multi-omics Integration
| Method Name | Data Types | Primary Function | Key Applications |
|---|---|---|---|
| Seurat Integration | scRNA-seq, scATAC-seq | Label transfer, co-embedding | Cell type annotation across modalities [55] |
| INSTINCT | spATAC-seq, multiple samples | Batch correction, domain identification | Spatial domain identification, multi-sample integration [58] |
| scPairing | Multiple single-cell modalities | Data generation and integration | Creating multiomics data from unimodal datasets [59] |
| PathVisio | Transcriptomics, proteomics, metabolomics | Pathway visualization | Multi-omics data visualization on biological pathways [60] |
| Chromatin Remodeling Analysis | ATAC-seq, ChIP-seq, RNA-seq, Hi-C | Regulatory network inference | Studying epigenetic remodeling by mutational synergy [57] |
Table 2: Common Multi-omics Integration Objectives in Translational Medicine
| Scientific Objective | Relevant Omics Combinations | Typical Applications |
|---|---|---|
| Subtype Identification | Genomics, transcriptomics, epigenomics | Patient stratification, disease classification [56] |
| Detect Disease-associated Molecular Patterns | Transcriptomics, epigenomics, proteomics | Biomarker discovery, mechanistic studies [56] |
| Understand Regulatory Processes | ATAC-seq, ChIP-seq, RNA-seq | Gene regulatory network inference, TF activity analysis [56] [57] |
| Diagnosis/Prognosis | Genomics, transcriptomics, metabolomics | Clinical prediction models, treatment response [56] |
| Drug Response Prediction | Transcriptomics, proteomics, epigenomics | Personalized treatment strategies, drug development [56] |
Experimental Protocols
Protocol 1: Integrating scRNA-seq and scATAC-seq Data Using Seurat
This protocol enables consistent annotation of both datasets with the same set of cell type labels and co-visualization of cells from scRNA-seq and scATAC-seq experiments [55].
Step-by-Step Methodology:
Data Preprocessing
- Process each modality independently using standard analysis pipelines [55].
- For scRNA-seq: Perform normalization, identify variable features, scale data, run PCA, and UMAP [55].
- For scATAC-seq: Add gene annotation information, run TF-IDF transformation, identify top features, run LSI dimensionality reduction, and UMAP [55].
Gene Activity Quantification
- Quantify transcriptional activity from scATAC-seq data using the
GeneActivity()function in Signac [55]. - Calculate ATAC-seq counts in the 2 kb-upstream region and gene body for all highly variable genes identified from the scRNA-seq dataset [55].
- Add gene activities as a new assay in the scATAC-seq object and normalize the data [55].
- Quantify transcriptional activity from scATAC-seq data using the
Anchor Identification
- Identify integration anchors using
FindTransferAnchors()with specific parameters for cross-modal integration [55]:- Set
reduction = "cca"to capture shared feature correlation structure. - Use reference (scRNA-seq) and query (scATAC-seq) datasets.
- Specify
reference.assay = "RNA"andquery.assay = "ACTIVITY".
- Set
- Identify integration anchors using
Label Transfer
- Transfer cell type annotations from scRNA-seq to scATAC-seq using
TransferData()[55]:- Provide annotations through the
refdataparameter. - Set
weight.reduction = pbmc.atac[["lsi"]]to use the ATAC-seq internal structure. - Include dimensions 2:30 from the LSI reduction.
- Provide annotations through the
- Transfer cell type annotations from scRNA-seq to scATAC-seq using
Validation and Evaluation
Protocol 2: Multi-omics Data Integration for Studying Chromatin Remodeling
This protocol provides an executable framework to study epigenetic remodeling induced by cooperating gene mutations and identify critical regulatory networks involved in disease [57].
Data Collection and Preprocessing:
Collect bulk next-generation sequencing (NGS) data for multiple genomic profiling approaches with biological replicates [57]:
- Chromatin accessibility (ATAC-seq)
- Chromatin activation states (ChIP-seq for H3K4me1, H3K4me3, H3K27ac)
- 3D chromatin interaction (Promoter capture HiC)
- Global gene expression (RNA-seq)
Process each dataset individually using specialized tools [57]:
- ATAC-seq: Process with FastQC, Bowtie2, MACS2 for peak calling
- ChIP-seq: Similar processing with additional normalization steps
- RNA-seq: Align with STAR, quantify expression with featureCounts or HTSeq
- pCHiC: Process with HiCUP, analyze interactions with CHiCAGO
Integrative Analysis:
- Perform differential analysis for each data type (e.g., using DESeq2 for RNA-seq) [57].
- Identify consensus peaks and regulatory elements across conditions.
- Link distal regulatory elements to target genes using pCHiC data.
- Correlate chromatin state changes with transcriptional changes.
- Identify master regulatory factors and networks driving phenotypic differences.
Computational System Requirements:
- Minimum 16 GB RAM and 12 CPU cores [57]
- R environment (v.4.1.3 or later) with packages: CHiCAGO, Seurat, DESeq2, DiffBind [57]
- Additional tools: FastQC, Bowtie2, Picard, MACS2, STAR, HiCUP [57]
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Multi-omics Experiments
| Reagent/Resource | Function/Purpose | Example Applications |
|---|---|---|
| 10x Genomics Multiome Kit | Simultaneous scRNA-seq and scATAC-seq profiling | Generating paired transcriptome and epigenome data from same cells [55] |
| Cell Barcoding Oligos | Sample multiplexing, batch effect reduction | Combining multiple samples in single experiments [54] |
| Unique Molecular Identifiers (UMIs) | Correcting PCR amplification biases | Quantitative scRNA-seq data interpretation [27] |
| Chromatin Antibodies (H3K4me1, H3K4me3, H3K27ac) | Histone modification profiling | Defining chromatin activation states in ChIP-seq [57] |
| Tn5 Transposase | Tagmentation of accessible chromatin regions | ATAC-seq library preparation [58] [57] |
| Template-Switching Oligos | cDNA amplification for full-length transcripts | Smart-seq2 protocols [27] |
Multi-omics Data Visualization
Effective visualization is crucial for interpreting multi-omics datasets. Below are the primary approaches:
Pathway-Based Visualization:
- Tools like PathVisio enable simultaneous visualization of different omics data types on pathway diagrams [60].
- Data preparation requires combining all measurements in one file with appropriate identifiers and system codes [60].
- Create intuitive visualizations using color gradients for expression values and rule-based coloring for different data types [60].
Metabolic Network Visualization:
- The Cellular Overview in Pathway Tools enables painting up to four omics datasets onto organism-scale metabolic charts [61].
- Different visual channels (color/thickness of reaction edges and metabolite nodes) represent distinct data types [61].
- Supports semantic zooming and animation for time-course data [61].
Downstream Analysis and Biological Interpretation
After successful integration of scRNA-seq with DNA and ATAC-seq data, several downstream analyses can reveal biologically significant insights:
Regulatory Network Inference:
- Identify transcription factors driving cellular identities and responses
- Link genetic variants to regulatory elements and target genes
- Construct gene regulatory networks using tools like SCENIC [54]
Spatial Domain Identification:
- Integrate spatial chromatin accessibility data (spATAC-seq) from multiple samples
- Identify spatially coordinated regulatory programs [58]
- Correlate spatial epigenetic patterns with tissue microstructure
Disease Mechanism Elucidation:
- Identify cooperative mutational effects on chromatin remodeling [57]
- Connect non-coding genetic variants to disease-associated genes
- Uncover cell-type specific dysregulation in complex diseases
The integration of scRNA-seq with DNA and ATAC-seq data provides unprecedented opportunities to understand cellular biology and disease mechanisms. The protocols outlined here enable researchers to connect genetic variation, epigenetic regulation, and transcriptional outcomes, offering a comprehensive framework for multi-omics analysis. As single-cell technologies continue to advance, these integration approaches will become increasingly essential for uncovering novel biological insights and advancing precision medicine initiatives.
Single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cellular heterogeneity and complex biological systems by enabling the transcriptomic profiling of individual cells [62]. This technological advancement is particularly transformative in drug discovery and development, where it facilitates the identification of novel therapeutic targets, enhances the credentialing of drug mechanisms of action, and aids in patient stratification through biomarker discovery [63] [40]. Two of the most critical analytical frameworks in scRNA-seq data interpretation are differential expression (DE) analysis, which identifies gene expression changes across conditions or cell types, and cell-cell communication (CCC) inference, which maps the signaling networks between different cell populations [64]. This application note provides detailed methodologies and protocols for implementing these functional analyses, framed within the context of pharmaceutical research and development.
Differential Expression Analysis in Drug Discovery
Analytical Challenges and Solutions
Differential expression analysis at single-cell resolution identifies genes that are statistically significantly expressed between distinct conditions, such as treated versus untreated cells, or between cell subpopulations. This analysis is pivotal for understanding drug mechanisms, identifying biomarkers, and discovering novel therapeutic targets [63] [65]. However, single-cell DE analysis presents unique computational challenges that differ substantially from bulk RNA-seq approaches.
Recent methodological evaluations have identified four major challenges in single-cell DE analysis, termed the "four curses": excessive zeros, normalization complexities, donor effects, and cumulative biases [66]. The high proportion of zero counts in scRNA-seq data arises from both biological phenomena (genuine absence of expression) and technical artifacts (so-called "dropout" events), complicating statistical modeling. Normalization challenges emerge from the need to correct for technical variations while preserving biological signals, particularly problematic when using count per million (CPM) approaches that convert unique molecular identifier (UMI)-based absolute counts to relative abundances, thereby erasing crucial quantitative information [66]. Donor effects (biological variability between samples) can confound results if not properly accounted for in experimental design and statistical modeling.
To address these challenges, a new statistical paradigm called GLIMES has been developed, which leverages UMI counts and zero proportions within a generalized Poisson/Binomial mixed-effects model [66]. This framework accounts for batch effects and within-sample variation while using absolute RNA expression rather than relative abundance, thereby improving sensitivity, reducing false discoveries, and enhancing biological interpretability.
Experimental Protocol for Differential Expression Analysis
Sample Preparation and scRNA-seq Library Generation
- Cell Isolation and Viability Assessment: Isolate single cells from tissue using enzymatic dissociation or mechanical disruption. For tissues difficult to dissociate (e.g., neuronal tissues), consider single-nuclei RNA-seq (snRNA-seq) [46]. Assess cell viability using trypan blue exclusion, ensuring >90% viability prior to library preparation.
- Single-Cell Capture and Barcoding: Utilize droplet-based systems (e.g., 10X Genomics Chromium) that employ microfluidics to encapsulate individual cells in droplets containing barcoded beads [46] [67]. Alternatively, use plate-based methods (e.g., SMART-Seq2) for full-length transcript coverage.
- Library Preparation and Sequencing: Reverse transcribe captured mRNA, amplify cDNA, and prepare sequencing libraries following manufacturer protocols. Sequence libraries to a minimum depth of 50,000 reads per cell to ensure adequate gene detection.
Computational Analysis Workflow
Quality Control and Preprocessing:
- Filter cells with low unique gene counts (<200 genes) or high mitochondrial content (>20%), indicating poor viability or apoptosis.
- Filter out genes detected in fewer than 10 cells to reduce noise in downstream analysis.
- For UMI-based data, retain raw counts without imputation to preserve quantitative information [66].
Normalization and Integration:
- Apply SCTransform (v2) for variance stabilizing transformation that regularizes Pearson residuals [66].
- For multi-sample experiments, perform integration using reciprocal PCA (RPCA) or Harmony to correct batch effects while preserving biological variation.
Differential Expression Testing:
- For comparing conditions within cell types, use mixed-effects models (e.g., GLIMES) that incorporate donor/sample identity as random effects [66].
- For identifying marker genes between cell types, use non-parametric Wilcoxon rank-sum tests.
- Apply multiple testing correction using Benjamini-Hochberg procedure to control false discovery rate (FDR < 0.05).
Table 1: Key Computational Tools for Differential Expression Analysis
| Tool | Methodology | Use Case | Advantages |
|---|---|---|---|
| GLIMES | Generalized Poisson/Binomial mixed-effects model | Condition-specific DE within cell types | Accounts for donor effects; uses absolute counts |
| Seurat | Wilcoxon rank-sum test, MAST | Marker gene identification | User-friendly; integrates with clustering |
| SCTransform | Regularized negative binomial regression | Data normalization and feature selection | Preserves biological variance; mitigates technical noise |
| SingleCellExperiment | Container for single-cell data | Framework for multiple DE methods | Flexible; compatible with Bioconductor ecosystem |
Cell-Cell Communication Analysis
Theoretical Framework and Applications
Cell-cell communication (CCC) analysis infers signaling interactions between different cell types based on the coordinated expression of ligand-receptor pairs [64]. In the context of drug discovery, mapping CCC networks helps elucidate disease mechanisms, identify novel therapeutic targets for disrupting pathogenic signaling, and understand how drugs modulate tissue microenvironment communication [63] [64].
A recent advancement in CCC analysis is the recognition of crosstalk between signaling pathways, where pathways activated by different ligand-receptor pairs may interact through shared signaling components [64]. This crosstalk can significantly impact signal fidelity (a pathway's capacity to prevent activation of its target by non-cognate signals) and specificity (a pathway's ability to avoid activating non-targets with its own signal). Understanding these regulatory concepts is essential for predicting drug effects on signaling networks.
Protocol for CCC Inference and Crosstalk Analysis
Experimental Design Considerations
- Sample Collection and Processing: Collect biologically relevant samples (e.g., tumor biopsies, PBMCs, tissue sections) with preservation of cell viability. Include appropriate controls and replicates (minimum n=3 per condition) to account for biological variability.
- Spatial Context Considerations: When possible, complement scRNA-seq with spatial transcriptomics or imaging data to validate inferred CCC, as physical proximity constraints enhance prediction accuracy [64] [62].
Computational Analysis Using SigXTalk
SigXTalk is a machine learning-based method that analyzes crosstalk in CCC using scRNA-seq data by quantifying signal fidelity and specificity [64]. The protocol implementation:
Input Data Preparation:
- Format scRNA-seq data as a count matrix with genes as rows and cells as columns.
- Provide cell type annotations derived from clustering and marker gene analysis.
- Optionally, specify target genes of interest; otherwise, SigXTalk uses differentially expressed genes per cluster.
CCC Network Reconstruction:
- Run CellChat to identify significantly active ligand-receptor pairs between cell populations [64].
- Filter interactions by expression thresholds (ligand > 0.1, receptor > 0.1 in respective cell populations).
Crosstalk Analysis:
- Construct a prior hypergraph skeleton using regulatory pathways from curated databases (e.g., NicheNet) [64].
- Train hypergraph neural network to predict pathway activation probabilities, aggregating information from neighbors on the hypergraph to incorporate higher-order regulatory interactions.
- Calculate pathway regulatory strength (PRS) using Random Forest regression of SSC-target pair activation levels against signal expression levels.
- Compute fidelity and specificity scores for each pathway within crosstalk modules.
Result Interpretation:
- Identify pathways with low fidelity as potential sources of off-target drug effects.
- Prioritize high-specificity pathways as candidates for targeted therapeutic intervention.
- Visualize crosstalk modules to understand signal integration and allocation mechanisms.
Table 2: Research Reagent Solutions for Functional Analysis
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| 10X Chromium Controller | Single-cell partitioning and barcoding | Optimize cell loading density (500-10,000 cells) |
| UMI Barcoded Beads | Molecular labeling for digital counting | Essential for quantitative expression analysis |
| CellChatDB | Curated ligand-receptor interaction database | Contains interactions with auxiliary components |
| NicheNet Prior Knowledge | Ligand-target signaling networks | Provides regulatory potential scores |
| SigXTalk Package | Crosstalk analysis in CCC | Requires Seurat or SingleCellExperiment objects |
Integrated Workflow for Drug Discovery Applications
Target Identification and Validation
The integration of DE and CCC analyses provides a powerful approach for target identification and validation in pharmaceutical research:
- Identify Disease-Associated Cell Subpopulations: Perform DE analysis to identify cell types or states expanded in disease conditions compared to healthy controls [63] [65].
- Characterize Dysregulated Signaling Pathways: Apply CCC inference to identify ligand-receptor pairs specifically active in disease microenvironments [64].
- Prioritize Therapeutic Targets: Integrate DE and CCC results to prioritize targets that are (i) specifically expressed in disease cell types, (ii) involved in pathogenic signaling pathways, and (iii) computationally predicted to have high fidelity and specificity [63].
- Experimental Validation: Design CRISPR-based functional genomics screens incorporating scRNA-seq readouts to validate target-disease associations and identify resistance mechanisms [63] [40].
Biomarker Discovery and Patient Stratification
ScRNA-seq enables biomarker discovery at unprecedented resolution by identifying cell-type-specific expression signatures associated with treatment response:
- Pre-treatment Biomarker Identification: Perform DE analysis on pre-treatment patient samples to identify gene expression signatures predictive of drug response [63] [68].
- Communication Network Biomarkers: Identify CCC patterns associated with resistance or sensitivity to therapy [64].
- Clinical Translation: Develop targeted gene expression panels measuring identified biomarkers for clinical implementation, potentially using more cost-effective bulk profiling approaches informed by single-cell discoveries [65].
Workflow for Integrated Analysis - This diagram illustrates the sequential steps for combining differential expression and cell-cell communication analyses in drug discovery applications.
CCC with Crosstalk - This diagram visualizes cell-cell communication pathways with crosstalk through shared signaling components, affecting signal fidelity and specificity.
The integration of differential expression analysis and cell-cell communication inference from scRNA-seq data provides a powerful framework for advancing drug discovery and development. By implementing the detailed protocols outlined in this application note, researchers can identify novel therapeutic targets with enhanced specificity, decipher drug mechanisms of action at cellular resolution, and develop biomarkers for patient stratification. As single-cell technologies continue to evolve, with improvements in spatial context preservation and multi-omics integration, these functional analyses will become increasingly central to pharmaceutical research, enabling more effective and targeted therapeutic interventions.
Solving scRNA-seq Challenges: Technical Noise, Batch Effects, and Data Integration
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the characterization of gene expression at unprecedented resolution, revealing cellular heterogeneity, identifying rare cell types, and illuminating developmental trajectories. However, the accurate interpretation of scRNA-seq data is critically dependent on addressing two major technical artifacts: dropout events and ambient RNA contamination.
Dropout events refer to the phenomenon where a gene is actively expressed in a cell but fails to be detected during sequencing, resulting in a false zero value in the expression matrix. This occurs due to the exceptionally low starting quantities of mRNA in individual cells and inefficiencies in cDNA synthesis and amplification [69]. Ambient RNA contamination arises in droplet-based scRNA-seq platforms when RNA molecules released from dead or dying cells in the cell suspension are co-encapsulated with intact cells and subsequently sequenced alongside the cell's native mRNA [70] [71]. This results in a background contamination profile that can obscure true biological signals.
Within the broader thesis of single-cell RNA sequencing data analysis research, this application note provides detailed protocols and frameworks for addressing these technical challenges, enabling researchers to extract more reliable biological insights from their experiments.
Understanding and Addressing Dropout Events
The Nature and Impact of Dropouts
Dropout events create a zero-inflated data structure that disproportionately affects lowly to moderately expressed genes. The probability of dropout is inversely correlated with true expression levels, with highly expressed genes less likely to be affected [69]. This technical noise can severely impact downstream analyses by obscuring gene-gene relationships, masking true cellular heterogeneity, and reducing the power to identify rare cell populations [69] [72]. Notably, transcriptional regulators, including many transcription factors, are often lowly expressed and therefore particularly vulnerable to dropout effects, potentially blinding analyses to key regulatory elements.
Performance Evaluation of Imputation Methods
A comprehensive evaluation of 11 imputation methods on 12 real datasets and 4 simulated datasets revealed critical insights into their performance characteristics [73]. The study assessed methods based on numerical recovery (ability to approximate true expression values), cell clustering consistency, and marker gene analysis.
Table 1: Performance Characteristics of Select Dropout Imputation Methods
| Method | Underlying Approach | Performance on Real Datasets | Performance on Simulated Datasets | Key Limitations |
|---|---|---|---|---|
| SAVER | Bayesian-based | Relatively good and stable performance in cell clustering; slight consistent improvement in numerical recovery | Higher errors due to statistical model assumptions | Tends to significantly underestimate expression values |
| scImpute | Mixture model & non-negative least squares | Improves clustering quality on some datasets | Performs well on data with collinearity | Can result in extremely large expression values; performs poorly with less collinearity |
| DCA | Deep autoencoder | Tends to overestimate expression values | Generally performs well, especially at high dropout rates | - |
| scVI | Deep generative model | Significant overestimation of expression values | Higher errors due to statistical model assumptions | Results in extremely large expression values |
| scScope | Deep learning | Significant underestimation of expression values | Excellent performance even at 90% dropout rates | Performs poorly on some real datasets |
| PBLR | Cell sub-population based bounded low-rank recovery | Effective in recovering dropouts and improving low-dimensional representation | Superior accuracy in recovering dropouts across multiple scenarios | Requires identification of cell sub-populations |
| GNNImpute | Graph attention network | Achieves low imputation error and high correlation with true values | Not specifically evaluated | Requires construction of cell-cell graph |
Performance varied substantially across different sequencing protocols. On 10x Genomics datasets, most methods explicitly improved corrupted data, while on Smart-Seq2/Smart-Seq datasets, many methods introduced additional noise with higher median errors [73]. Surprisingly, some imputation methods had a negative effect on cell clustering consistency compared to raw count data, particularly on datasets with clear intrinsic clustering structures [73].
Protocol for Dropout Imputation Using GNNImpute
GNNImpute represents a recent advancement leveraging graph attention networks to address dropouts [74]. Below is a detailed protocol for its implementation:
Input Requirements: A raw count matrix (cells × genes) with zeros representing potential dropout events.
Step 1: Data Preprocessing
- Filter the raw count matrix to remove cells with fewer than 200 total counts and genes detected in fewer than 3 cells.
- Remove cells with overexpression of mitochondrial genes, indicative of poor cell quality or apoptosis.
- Filter cells with extremely high total counts, potentially representing multiplets or counting errors.
- Normalize the filtered matrix so each cell has the same total count (median normalization across cells).
Step 2: Construction of Cell-Cell Graph
- Perform dimensionality reduction on the preprocessed expression matrix using Principal Component Analysis (PCA), retaining the top 50 principal components.
- Calculate pairwise Euclidean distances between all cells in the PCA-reduced space.
- Construct a k-nearest neighbor graph (default k=5) where nodes represent cells and edges connect each cell to its k most similar neighbors.
Step 3: Graph Attention Imputation
- Implement a neural network with an encoder-decoder architecture.
- The encoder consists of two graph attention layers that aggregate information from first- and second-level neighbors using multi-head attention mechanisms.
- The decoder consists of two fully connected layers that reconstruct the imputed expression matrix.
- Train the model using the preprocessed expression matrix as both input and target, optimizing parameters to minimize the difference between output and input.
Step 4: Output and Validation
- The model outputs a denoised, imputed count matrix.
- Validate imputation quality by assessing clustering coherence and comparing with known cell-type markers.
Diagram 1: GNNImpute workflow for dropout imputation in scRNA-seq data
Alternative Approach: Leveraging Dropout Patterns
Contrary to conventional approaches that treat dropouts as noise to be removed, emerging methodologies leverage dropout patterns as valuable biological signals. The co-occurrence clustering algorithm utilizes binarized expression data (0 for non-detection, 1 for detection) to identify cell populations [75]. This approach identifies genes with correlated dropout patterns across cells, which often correspond to functional pathways and can define meaningful cell subtypes without relying on highly variable genes.
Understanding and Addressing Ambient RNA Contamination
Ambient RNA contamination originates from extracellular mRNA molecules in the cell suspension that are co-encapsulated with cells during the droplet generation process [71]. These molecules typically derive from stressed, apoptotic, or necrotic cells that have released their contents into the solution. In droplet-based systems, this contamination affects both empty droplets and cell-containing droplets, with the latter capturing a mixture of endogenous and ambient transcripts [70].
The consequences of ambient RNA contamination include:
- Erosion of cell type identity: Cell type-specific markers from abundant cell types appear to be expressed in unrelated cell types [71].
- Reduced cluster resolution: Distinct cell populations appear more similar, potentially merging separate populations [76].
- Biological misinterpretation: Appearing expression of genes inconsistent with cell type may lead to incorrect biological conclusions [70].
- Masking of rare cell types: Rare cell populations may be obscured by contamination signatures from dominant populations.
The extent of contamination varies substantially across experimental protocols. Comparative analyses have shown that 10X Chromium typically exhibits the lowest levels of contamination, while CEL-seq2 demonstrates the highest [71].
Performance Evaluation of Decontamination Methods
Multiple computational approaches have been developed to address ambient RNA contamination, each with distinct methodologies and performance characteristics.
Table 2: Performance Characteristics of Ambient RNA Removal Methods
| Method | Underlying Approach | Key Strengths | Key Limitations |
|---|---|---|---|
| DecontX | Bayesian model with multinomial distributions | Accurate estimation of contamination levels; individual cell estimates | Requires cell population labels for optimal performance |
| SoupX | Estimates contamination from empty droplets | Does not require cell clustering; simple implementation | Contamination fraction estimation can be complex |
| CellBender | Deep generative model | Performs both cell-calling and ambient RNA removal; comprehensive solution | Computationally intensive; requires GPU for efficiency |
| EmptyNN | Neural network classifier | Identifies and removes empty droplets | Failed to call cells in certain tissue types |
| DIEM | Expectation-Maximization algorithm | Identifies debris on cell-by-cell basis | Multiple user-defined thresholds introduce subjectivity |
DecontX demonstrates particularly robust performance, accurately quantifying and removing contamination in diverse experimental contexts. In a human-mouse mixture dataset, DecontX estimates strongly correlated with the actual proportion of cross-species transcripts (R = 0.99) [71]. Applied to PBMC datasets, it effectively removed aberrant expression of marker genes in inappropriate cell types, enhancing biological interpretability [71].
Protocol for Ambient RNA Removal Using DecontX
DecontX employs a Bayesian framework to deconvolute the observed expression counts into native and contamination components [71]. The detailed protocol follows:
Input Requirements: A raw count matrix (genes × cells) and optional cell cluster labels.
Step 1: Model Formulation
- Assume the observed count matrix for each cell follows a mixture of two multinomial distributions:
- Native distribution: Transcripts originating from the cell's actual biological population
- Contamination distribution: Ambient transcripts originating from all other cell populations
- Define parameters:
- θj: Proportion of counts in cell j derived from native distribution (Beta distributed)
- φk: Native expression distribution for cell population k (Multinomial)
- η_k: Contamination distribution for cell population k (Multinomial)
Step 2: Variational Inference
- Approximate the posterior distributions of latent variables using variational inference for computational efficiency.
- For each cell j, estimate the contamination proportion θj and the distributions φk and η_k.
- The contamination distribution for each cell population is modeled as a weighted combination of all other cell population distributions.
Step 3: Decontamination
- Deconvolute the observed count matrix into two matrices:
- Native counts matrix: Estimated counts from biological expression
- Contamination counts matrix: Estimated counts from ambient RNA
- Use the native counts matrix for all downstream analyses.
Step 4: Validation
- Check for reduction in inappropriate marker gene expression across cell types.
- Verify that genuine cell-type-specific markers are preserved.
- Assess cluster separation in low-dimensional embeddings.
Diagram 2: DecontX workflow for ambient RNA removal in scRNA-seq data
Experimental Optimization to Minimize Ambient RNA
Beyond computational correction, experimental optimizations can significantly reduce ambient RNA contamination at its source [77]:
- Cell Loading Optimization: Precise control of cell loading rates significantly impacts contamination levels. Overloading increases multiplet rates, while underloading reduces cell capture efficiency.
- Cell Fixation: Appropriate fixatives can stabilize cells during processing, reducing RNA leakage without significantly compromising RNA quality.
- Microfluidic Dilution: Adjusting buffer-to-cell ratios in microfluidic systems can reduce co-encapsulation of ambient RNA.
- Nuclei vs. Whole Cell Preparation: For tissues prone to dissociation-induced stress, nuclei isolation may reduce ambient RNA, though this comes with transcriptional bias.
Integrated Workflow and Decision Framework
Strategic Application of Correction Methods
The decision to apply dropout imputation, ambient RNA removal, both, or neither depends on multiple factors, including experimental system, data quality, and analytical goals. The following framework provides guidance:
Apply both corrections when:
- Working with tissues known for high cell death or stress (e.g., solid tumors)
- Studying rare cell populations where signal-to-noise ratio is critical
- Analyzing data with clear evidence of contamination (e.g., inappropriate marker expression)
Apply only ambient RNA removal when:
- Dropout rates are low (e.g., high sequencing depth)
- Preserving true zeros is critical for biological interpretation
Apply only dropout imputation when:
- Working with sorted populations or high-viability cells minimizing ambient RNA
- Focusing on improving gene-gene relationships or trajectory inference
Apply neither correction when:
- Analyzing high-quality datasets with minimal technical artifacts
- Utilizing methods that explicitly leverage dropout patterns [75]
Quality Control Metrics
Systematic quality assessment is essential before and after applying correction methods:
Pre-correction QC:
- Barcode rank plots: Assess the separation between cell-containing and empty droplets [70]
- Contamination-focused metrics: Utilize geometric or statistical metrics to quantify contamination levels [77]
- Marker gene specificity: Check for inappropriate expression of cell-type-specific markers
Post-correction QC:
- Cluster coherence: Evaluate silhouette coefficients and other clustering metrics [73]
- Biological plausibility: Verify that known biological relationships are preserved or enhanced
- Method-specific diagnostics: Review method-generated diagnostics (e.g., estimated contamination fractions in DecontX)
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Addressing Technical Artifacts
| Tool/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Decontamination Software | DecontX, SoupX, CellBender | Estimate and remove ambient RNA contamination | Post-sequencing data processing for droplet-based methods |
| Imputation Algorithms | GNNImpute, PBLR, SAVER, DCA | Predict and correct for dropout events | Recovery of missing values in sparse scRNA-seq matrices |
| Quality Control Tools | EmptyNN, DropletQC, CellBender | Distinguish cell-containing from empty droplets | Preprocessing and cell calling in droplet-based data |
| Clustering Methods | SC3, PhenoGraph, Seurat | Identify cell populations and subtypes | Downstream analysis after data correction |
| Experimental Solutions | Nuclei Isolation Kits, Cell Fixation Reagents | Reduce RNA leakage and ambient contamination | Sample preparation for challenging tissues |
Addressing technical artifacts in scRNA-seq data through thoughtful application of dropout imputation and ambient RNA removal methods is essential for extracting biologically meaningful insights. The protocols and frameworks presented here provide researchers with practical guidance for implementing these corrections while understanding their limitations and appropriate contexts.
Performance evaluations consistently show that method performance is dataset-dependent, with no single approach universally superior across all scenarios [73] [78]. This underscores the importance of method benchmarking for specific experimental systems and analytical goals. Furthermore, the integration of experimental optimizations with computational corrections represents the most robust strategy for managing technical artifacts.
As the field advances, we anticipate increased method specialization for particular biological contexts, improved integration of external information sources [72], and more sophisticated approaches that simultaneously address multiple technical artifacts. Through the careful application of these evolving methodologies, researchers can significantly enhance the reliability and biological relevance of their single-cell transcriptomic studies.
In single-cell RNA sequencing (scRNA-seq) data analysis, the percentage of mitochondrial RNA counts (pctMT) has traditionally served as a key quality control metric for identifying dying or low-viability cells [79]. Conventional bioinformatics pipelines routinely filter out cells exhibiting high pctMT, based on the established correlation between elevated mitochondrial RNA content and dissociation-induced stress or necrosis [79]. However, emerging evidence challenges this universal application, particularly in disease contexts such as cancer, where elevated pctMT may represent genuine biological signals rather than technical artifacts [79]. This protocol provides a structured framework for distinguishing biologically relevant mitochondrial signals from cell death artifacts in scRNA-seq data, enabling researchers to preserve functionally important cell populations that would otherwise be excluded by standard quality control filters.
Background and Significance
The Dual Interpretation of Mitochondrial RNA Content
Mitochondrial RNA content in scRNA-seq data presents a complex interpretive challenge. While high pctMT values often indicate compromised cellular integrity, they can also reflect genuine biological states characterized by elevated metabolic activity or mitochondrial dysfunction [79] [80]. Malignant cells frequently exhibit naturally higher baseline mitochondrial gene expression compared to their nonmalignant counterparts, potentially due to increased mitochondrial DNA copy number or metabolic reprogramming [79]. Analysis of 441,445 cells across 134 cancer patients revealed that malignant cells consistently show significantly higher pctMT than nonmalignant cells in the tumor microenvironment, with 72% of samples demonstrating this pattern [79].
Mitochondrial RNA Biology
The mitochondrial transcriptome includes 13 protein-coding genes, 2 ribosomal RNAs (rRNAs), 22 transfer RNAs (tRNAs), and various noncoding RNAs encoded by the mitochondrial genome [81] [82]. These molecules participate in critical cellular processes including energy production, metabolism, and signaling. Nuclear-encoded noncoding RNAs such as microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs) also regulate mitochondrial function through anterograde-retrograde communication between the nucleus and mitochondria [81].
Table 1: Key Mitochondrial RNA Types and Functions
| RNA Type | Origin | Key Functions | Example |
|---|---|---|---|
| Protein-coding mRNAs | mtDNA | Encode subunits of oxidative phosphorylation complexes | MT-ND1, MT-CO1 |
| Transfer RNAs (tRNAs) | mtDNA | Mitochondrial protein synthesis | MT-TL1, MT-TS1 |
| Ribosomal RNAs (rRNAs) | mtDNA | Mitochondrial ribosome assembly | MT-RNR1, MT-RNR2 |
| microRNAs (mitomiRs) | mtDNA/nDNA | Post-transcriptional regulation of mitochondrial genes | miR-181c, miR-2392 |
| Long noncoding RNAs | mtDNA/nDNA | RNA stabilization, regulatory functions | lncND5, LIPCAR |
| Circular RNAs | mtDNA/nDNA | Protein binding, miRNA sponges | circRNA SCAR, circPUM1 |
Quality Control Assessment Framework
Establishing Context-Appropriate Thresholds
Traditional pctMT filtering thresholds (typically 10-20%) were primarily established using healthy tissues and may be overly stringent for certain biological contexts [79]. Cancer studies reveal that applying a standard 15% pctMT filter would eliminate 10-50% of malignant cells across various cancer types, potentially discarding biologically relevant populations [79]. The framework below outlines a systematic approach for evaluating mitochondrial content.
Quantitative Guidelines for pctMT Interpretation
Table 2: Interpretation Framework for Mitochondrial RNA Content
| Metric | Traditional Approach | Recommended Refinement | Interpretation Guidelines |
|---|---|---|---|
| pctMT Threshold | Apply uniform threshold (10-20%) across all cells [79] | Establish cell-type-specific thresholds | Malignant cells often exhibit 1.5-2x higher pctMT than nonmalignant counterparts [79] |
| Stress Association | Assume high pctMT indicates dissociation stress | Quantify using dissociation-induced stress signatures | Weak correlation (point biserial coefficient <0.3) suggests biological origin [79] |
| Cell Viability | Exclude high pctMT cells as non-viable | Integrate additional viability metrics (MALAT1, nuclear debris) | High pctMT cells passing other QC metrics likely represent viable states [79] |
| Biological Validation | Not typically performed | Compare with spatial transcriptomics or bulk RNA-seq | Spatial data can confirm high mitochondrial gene expression in viable tissue regions [79] |
Experimental Protocols
Protocol 1: Dissociation-Induced Stress Assessment
Purpose: To determine whether elevated pctMT values result from technical artifacts during tissue dissociation.
Materials:
- Processed scRNA-seq data (post-initial QC)
- Dissociation-induced stress gene signatures [79]
- Computational environment (R/Python) with single-cell analysis tools
Methodology:
- Construct Meta Stress Signature: Compile genes consistently identified as dissociation stress markers across multiple studies (e.g., from O'Flanagan et al., Machado et al., and van den Brink et al.) [79].
- Calculate Stress Scores: Using the AddModuleScore function in Seurat or equivalent, compute dissociation stress scores for each cell.
- Compare Distributions: Assess stress scores between HighMT and LowMT populations within both malignant and nonmalignant compartments.
- Statistical Evaluation: Apply Mann-Whitney U test to evaluate significance, with effect size calculation (point biserial coefficient).
- Interpretation: Weak correlations (<0.3) between pctMT and stress scores suggest biological rather than technical origins of high pctMT.
Protocol 2: Spatial Validation of Mitochondrial RNA Expression
Purpose: To confirm the viability and biological significance of high pctMT cells using spatial transcriptomics.
Materials:
- Visium HD spatial transcriptomics data
- Corresponding scRNA-seq dataset
- Image analysis software
Methodology:
- Region Identification: Identify tissue regions with elevated expression of mitochondrial-encoded genes in spatial data.
- Viability Assessment: Evaluate morphological features in H&E-stained adjacent sections to confirm cellular viability in high-mitochondrial RNA regions.
- Integration Analysis: Map scRNA-seq clusters to spatial locations to determine if high pctMT cells localize to viable tissue regions.
- Correlation Analysis: Assess concordance between mitochondrial gene expression patterns in scRNA-seq and spatial transcriptomics data.
Protocol 3: Functional Characterization of High pctMT Cells
Purpose: To determine whether high pctMT cells represent metabolically active populations with potential clinical relevance.
Materials:
- scRNA-seq data with cell type annotations
- Metabolic pathway gene sets
- Clinical metadata (when available)
Methodology:
- Pathway Analysis: Perform gene set enrichment analysis on HighMT versus LowMT cells using metabolic pathways.
- Xenobiotic Metabolism Assessment: Evaluate expression of drug metabolism genes in high pctMT populations.
- Clinical Correlation: Associate high pctMT populations with patient outcomes or treatment responses where data available.
- Trajectory Analysis: Utilize pseudotime tools (Monocle2, VECTOR) to determine if high pctMT cells represent transitional states.
Analytical Workflow Implementation
Comprehensive Decision Framework
The diagram below outlines the complete analytical workflow for distinguishing biological signals from technical artifacts in mitochondrial RNA analysis.
Mitochondrial Dysfunction in Disease Contexts
Research across multiple disease models confirms the importance of preserving high pctMT cells for biological discovery. In microtia chondrocytes, single-cell RNA sequencing revealed mitochondrial dysfunction characterized by increased ROS production, decreased membrane potential, and altered mitochondrial structure [80]. In amyotrophic lateral sclerosis (ALS), transcriptomic analyses of motor neurons identified early mitochondrial impairments as shared pathological mechanisms across FUS- and TARDBP-ALS mutations [83]. These findings underscore the value of retaining high pctMT cells when they represent genuine biological phenomena rather than technical artifacts.
Research Reagent Solutions
Table 3: Essential Research Reagents for Mitochondrial RNA Analysis
| Reagent/Technology | Provider | Function in Mitochondrial Analysis | Key Applications |
|---|---|---|---|
| 10x Genomics Chromium | 10x Genomics | Single-cell partitioning and barcoding | High-throughput scRNA-seq with mitochondrial transcript capture |
| MitoCarta3.0 | Broad Institute | Curated inventory of 1,136 human mitochondrial genes | Reference for mitochondrial gene set scoring and pathway analysis |
| MAESTER | Public protocol | Enrichment of mitochondrial mutations from cDNA libraries | Detection of mtDNA variants from high-throughput scRNA-seq data |
| mtscATAC-seq | Public protocol | Combined chromatin accessibility and mitochondrial genome sequencing | Mitochondrial clonality assessment with epigenetic profiling |
| ASAP-seq/DOGMA-seq | Public protocol | Multimodal profiling (transcriptome, chromatin, surface protein, mtDNA) | Comprehensive single-cell analysis with mitochondrial genotyping |
| mgatk/maegatk | Open-source software | Variant calling and heteroplasmy quantification from mtDNA/RNA data | Genotyping software for mitochondrial mutation analysis |
Rigid application of pctMT filtering thresholds risks eliminating biologically significant cell populations, particularly in disease contexts such as cancer. The framework presented herein enables discrimination between technical artifacts and genuine biological signals through multi-modal assessment of dissociation stress, metabolic activity, and spatial localization. Implementation of these protocols will enhance detection of functionally relevant cell states characterized by elevated mitochondrial RNA content, potentially advancing discovery in disease mechanisms and therapeutic development.
Batch Effect Correction and Harmonization of Multi-Sample Datasets
The advent of single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the exploration of cellular heterogeneity at an unprecedented resolution. However, as researchers increasingly combine datasets from different experiments, times, or technologies to increase statistical power and discovery potential, they encounter the significant challenge of batch effects. These are technical variations that are introduced not due to biological differences but from factors such as different laboratory conditions, personnel, sequencing platforms, or reagent batches [84]. When integrating multiple scRNA-seq datasets, these technical artifacts can confound true biological signals, leading to misleading conclusions in downstream analyses such as cell type identification, differential expression analysis, and trajectory inference [85] [86].
The need for effective batch effect correction is particularly acute in large-scale "atlas" projects that aim to combine public datasets with substantial technical and biological variation, including multiple organs and developmental stages [85]. The fundamental goal of batch effect correction is to remove these non-biological technical variations while preserving the genuine biological signals that researchers seek to understand. This balance is delicate; under-correction leaves batch effects that can mask true biological differences, while over-correction can erase meaningful biological variation and lead to false discoveries [86]. The integration of datasets across different systems—such as species, organoids and primary tissue, or different scRNA-seq protocols—presents particularly substantial challenges that require advanced correction methodologies [85].
Batch effects in scRNA-seq data arise from multiple technical sources throughout the experimental workflow. These include library preparation protocols (e.g., Smart-seq2, 10X Genomics 3' or 5' kits), sequencing platforms (Illumina, PacBio, Oxford Nanopore), reagent lots, personnel differences, and laboratory conditions [84] [86]. Even when samples are processed using the same nominal protocol, subtle variations in execution can introduce systematic technical differences that manifest as batch effects. These technical factors collectively create variations in gene expression measurements that are unrelated to the biological phenomena under investigation.
Biological Consequences in Downstream Analysis
The impact of uncorrected batch effects permeates nearly every aspect of single-cell data analysis. In cell type identification and clustering, batch effects can cause cells of the same type to appear distinct or cells of different types to appear similar, leading to incorrect cell type annotations [86]. For differential expression analysis, batch effects can create false positives or mask truly differentially expressed genes, particularly when batch is confounded with biological conditions of interest. In trajectory inference, batch effects can distort the inferred developmental paths, while in cell-cell communication analysis, they can create artificial signaling patterns or obscure real ones [86]. These distortions become particularly problematic when integrating data across different biological systems, such as human and mouse samples, or between organoids and primary tissues, where the biological differences themselves are of primary interest [85].
Comparative Analysis of Batch Correction Methods
Multiple computational methods have been developed to address batch effects in scRNA-seq data, each with distinct theoretical foundations, input requirements, and correction strategies. These methods can be broadly categorized based on whether they correct the count matrix directly or instead correct a lower-dimensional embedding or the k-nearest neighbor (k-NN) graph derived from the data [84]. Methods that modify the count matrix (e.g., Combat, ComBat-seq, MNN, Seurat) directly adjust gene expression values, while those that modify embeddings (e.g., Harmony, LIGER, SCVI) or graphs (e.g., BBKNN) affect downstream analyses that rely on these structures without altering the original counts [84].
Table 1: Comparison of scRNA-seq Batch Effect Correction Methods
| Method | Input Data | Correction Object | Key Algorithm | Output |
|---|---|---|---|---|
| Harmony | Normalized count matrix | Embedding | Soft k-means with linear batch correction within clusters | Corrected embedding |
| BBKNN | k-NN graph | k-NN graph | UMAP on merged neighborhood graph | Corrected k-NN graph |
| ComBat | Normalized count matrix | Count matrix | Empirical Bayes-linear correction | Corrected count matrix |
| ComBat-seq | Raw count matrix | Count matrix | Negative binomial regression | Corrected count matrix |
| LIGER | Normalized count matrix | Embedding | Quantile alignment of factor loadings | Corrected embedding |
| MNN | Normalized count matrix | Count matrix | Mutual nearest neighbors-linear correction | Corrected count matrix |
| SCVI | Raw count matrix | Embedding | Variational autoencoder modeling batch effects | Corrected count matrix & embedding |
| Seurat | Normalized count matrix | Embedding | Aligning canonical basis vectors | Corrected count matrix |
Performance Evaluation of Correction Methods
Recent benchmarking studies have evaluated the performance of these methods across various datasets and integration scenarios. A 2025 comparison of eight widely used methods found that many introduce measurable artifacts during the correction process [84]. Specifically, MNN, SCVI, and LIGER performed poorly in these tests, often altering the data considerably, while ComBat, ComBat-seq, BBKNN, and Seurat introduced artifacts that could be detected in their testing setup [84]. Notably, Harmony was the only method that consistently performed well across all evaluations, making it the recommended choice for standard batch correction scenarios [84].
However, for datasets with substantial batch effects—such as those integrating across species, between organoids and primary tissue, or across different technologies—even these methods may struggle. A 2025 study focusing on such challenging integration scenarios found that conditional variational autoencoder (cVAE)-based methods with VampPrior and cycle-consistency constraints (sysVI) showed particular promise for these difficult cases [85]. This approach demonstrated improved integration across systems while better preserving biological signals for downstream interpretation of cell states and conditions compared to existing methods [85].
Table 2: Performance Characteristics of Batch Correction Methods
| Method | Batch Removal Effectiveness | Biological Preservation | Overcorrection Risk | Recommended Use Cases |
|---|---|---|---|---|
| Harmony | High | High | Low | Standard batch integration |
| Seurat | Moderate-High | Moderate | Moderate | Standard batch integration |
| sysVI | High | High | Low | Cross-system integration |
| ComBat/ComBat-seq | Moderate | Moderate | High | Mild technical batch effects |
| SCVI | Moderate | Moderate | High | Large dataset integration |
| MNN | Moderate | Low | High | Not recommended |
| LIGER | Moderate | Low | High | Not recommended |
The RBET Framework: A Novel Approach for Evaluating Batch Correction
The Challenge of Overcorrection
A significant challenge in batch effect correction is the risk of overcorrection—the removal of true biological variation along with technical noise. This problem has been difficult to quantify using existing evaluation metrics such as kBET or LISI, which focus primarily on batch mixing but lack sensitivity to biological information loss [86]. Overcorrection can lead to false biological discoveries, such as the erroneous merging of distinct cell types or the artificial splitting of homogeneous populations [86].
Principles of the RBET Framework
The Reference-informed Batch Effect Testing (RBET) framework, introduced in 2025, addresses this limitation by leveraging the expression patterns of reference genes (RGs)—typically housekeeping genes with stable expression across various cell types and conditions [86]. The fundamental assumption underlying RBET is that properly integrated data should show no batch effects on these reference genes, both locally and globally. The framework consists of two main steps: (1) selection of appropriate reference genes specific to each dataset, and (2) detection of batch effects on these reference genes in the integrated dataset using maximum adjusted chi-squared (MAC) statistics [86].
In comprehensive evaluations, RBET demonstrated superior performance in detecting batch effects while maintaining awareness of overcorrection. Unlike other metrics, RBET values show a characteristic biphasic response during overcorrection: initially decreasing as true batch effects are removed, then increasing as biological signal is erased [86]. This unique property makes RBET particularly valuable for selecting appropriate correction strengths and comparing different batch correction methods.
Experimental Validation of RBET
When applied to real datasets, RBET has proven effective at identifying batch correction methods that preserve biological truth. In an analysis of pancreas data with three technical batches, RBET correctly identified Seurat as the best-performing method, resulting in superior cluster quality (as measured by Silhouette Coefficient) and higher accuracy in cell type annotation compared to methods favored by other metrics [86]. This demonstrates RBET's practical utility for ensuring biologically meaningful integration results.
Experimental Protocols for Batch Effect Correction
Pre-correction Quality Control and Normalization
Prior to batch effect correction, proper quality control and normalization are essential prerequisites:
- Quality Control Metrics: Filter cells based on unique gene counts (500-5000 genes/cell), total UMI counts, and mitochondrial percentage (typically <10-20%).
- Doublet Detection: Use algorithms like DoubletFinder or scDblFinder to identify and remove multiplets.
- Normalization: Apply standard normalization methods such as SCTransform (Seurat) or log-normalization (10,000 counts/cell).
- Feature Selection: Identify highly variable genes (2000-5000 genes) for downstream integration.
Implementation of Harmony for Batch Correction
Harmony operates on principal component analysis (PCA) embeddings and is implemented as follows:
- Input Preparation: Standard preprocessing and PCA on normalized count matrix.
- Parameter Settings:
theta: Diversity clustering penalty (default: 2)lambda: Ridge regression penalty (default: 1)max_iter: Maximum iterations (default: 10)
- Integration: Run Harmony integration using batch covariates.
- Downstream Analysis: Use Harmony embeddings for clustering, UMAP visualization, and trajectory analysis.
Implementation of sysVI for Substantial Batch Effects
For challenging integration scenarios with substantial batch effects (e.g., cross-species, technology integration):
- Architecture: Conditional variational autoencoder (cVAE) with VampPrior and cycle-consistency constraints.
- Input: Normalized count matrices from multiple systems.
- Training: Joint training with cycle-consistency loss to preserve biological variation.
- Output: Integrated embedding that removes system-specific technical effects while preserving cross-system biological signals.
Post-correction Evaluation Protocol
After applying batch correction, comprehensive evaluation is essential:
- Visual Assessment: Examine UMAP/t-SNE plots for batch mixing and biological separation.
- Quantitative Metrics: Calculate RBET, LISI, or kBET scores.
- Biological Validation:
- Check preservation of known cell type markers
- Verify consistency of biological patterns with prior knowledge
- Assess cluster purity using Silhouette Width
- Differential Expression: Confirm that known differentially expressed genes remain detectable.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for scRNA-seq Batch Correction
| Tool/Resource | Function | Application Context |
|---|---|---|
| 10X Genomics Chromium | Single-cell partitioning & barcoding | Generating 3' or 5' scRNA-seq data |
| Harmony R Package | Batch effect correction | Integrating multiple datasets with mild-moderate batch effects |
| sysVI Python Package | Substantial batch effect correction | Cross-system integration (species, technologies) |
| Seurat R Toolkit | Single-cell analysis & integration | End-to-end analysis with built-in correction methods |
| SCANPY Python Toolkit | Single-cell analysis & integration | Python-based analysis with multiple integration options |
| RBET Evaluation Framework | Batch correction assessment | Quantifying correction success with overcorrection awareness |
| Cell Ranger Pipeline | Sequencing data processing | Processing 10X Genomics data from raw sequences to count matrices |
Experimental Design Considerations
Proper experimental design can minimize batch effects from the outset:
- Biological Replicates: Include multiple biological replicates (not just technical replicates) to enable proper statistical testing [87].
- Randomization: Process samples from different conditions randomized across batches.
- Reference Samples: Include control or reference samples across batches to monitor technical variation.
- Balanced Design: Ensure conditions of interest are represented across multiple batches to avoid confounding.
Workflow Visualization
Batch Effect Correction Workflow
RBET Evaluation Framework
Batch effect correction remains a critical step in the analysis of multi-sample scRNA-seq datasets, particularly as the field moves toward larger atlas-level integrations. The emerging consensus recommends Harmony for standard integration tasks [84], while sysVI shows promise for more challenging cross-system integrations [85]. The development of evaluation frameworks like RBET that are sensitive to overcorrection represents a significant advance in ensuring that batch correction methods preserve biologically meaningful signals [86].
Looking forward, several areas require continued development: methods that can better handle substantial batch effects across different biological systems, approaches that scale to the millions of cells now being generated, and frameworks that provide clearer guidance on method selection for specific data scenarios. Furthermore, as multi-omic single-cell technologies mature, developing integration methods that can simultaneously correct batch effects across different data modalities will become increasingly important.
By following the protocols and recommendations outlined in this document, researchers can implement effective batch correction strategies that enable robust biological insights from integrated single-cell datasets while minimizing the risks of both under-correction and over-correction.
Optimizing Computational Performance for Large-Scale Data
The scale and complexity of data generated by single-cell RNA sequencing (scRNA-seq) technologies present substantial computational challenges. As researchers embark on projects involving thousands to millions of cells, the need for efficient processing, analysis, and interpretation pipelines becomes paramount. This application note provides detailed protocols and strategic guidance for optimizing computational performance in large-scale scRNA-seq studies, framed within a comprehensive thesis on single-cell data analysis. We focus on practical solutions that balance analytical accuracy with computational feasibility, enabling researchers to extract meaningful biological insights from massive datasets without prohibitive resource requirements.
Key Computational Bottlenecks in scRNA-seq Analysis
Large-scale scRNA-seq experiments generate data with distinctive computational characteristics that challenge conventional analysis approaches. The primary bottlenecks include:
Data Volume and Sparsity: Droplet-based scRNA-seq protocols can profile tens of thousands of cells in a single experiment, generating matrices with millions of rows (genes) and columns (cells) where most entries are zeros [27]. This sparsity necessitates specialized algorithms for efficient storage and computation.
High-Dimensional Space: The intrinsic high-dimensional nature of gene expression data (typically 20,000-30,000 dimensions) requires dimensionality reduction before most downstream analyses, creating computational bottlenecks in matrix operations and neighbor finding [88].
Iterative Analytical Processes: Clustering, trajectory inference, and integration often involve iterative algorithms that must be repeated with different parameters, multiplying computational demands [89].
Table 1: Computational Bottlenecks in Large-Scale scRNA-seq Analysis
| Bottleneck Category | Specific Challenges | Impact on Analysis |
|---|---|---|
| Memory Requirements | Storage of large sparse matrices; Loading full datasets into RAM | Limits simultaneous processing of multiple samples; Requires specialized data structures |
| Processing Power | High-dimensional calculations; Matrix factorization; Graph construction | Extends analysis time from hours to days; Requires high-performance computing (HPC) |
| Algorithmic Complexity | Nearest-neighbor search in high dimensions; Iterative clustering optimization | Creates scalability issues with increasing cell numbers |
| Data Integration | Batch correction across multiple datasets; Reference mapping | Requires sophisticated algorithms to maintain biological variation while removing technical artifacts |
Strategic Optimization Approaches
Feature Selection for Enhanced Performance
Feature selection represents one of the most effective strategies for improving computational performance while maintaining biological relevance. By focusing analysis on the most informative genes, researchers can significantly reduce dimensionality and enhance algorithm efficiency.
A recent comprehensive benchmark study evaluated over 20 feature selection methods and demonstrated that highly variable gene selection consistently improves integration performance and computational efficiency [23]. The study revealed that selecting 2,000-3,000 highly variable features typically optimizes the trade-off between biological preservation and computational requirements, with diminishing returns beyond this range.
Protocol: Highly Variable Feature Selection
- Input: Raw or normalized count matrix (cells × genes)
- Calculation: Compute mean expression and dispersion for each gene
- Selection: Retain genes with highest dispersion relative to their mean
- For Scanpy:
sc.pp.highly_variable_genes(adata, n_top_genes=3000) - For Seurat:
FindVariableFeatures(selection.method = "vst", nfeatures = 3000)
- For Scanpy:
- Validation: Ensure selected features capture known cell-type markers
- Downstream Application: Use selected features for dimensionality reduction, clustering, and integration
Batch-aware feature selection methods further enhance performance when integrating datasets from different sources by identifying features with consistent biological variation across batches [23].
Efficient Data Structures and Matrix Representations
The sparse nature of scRNA-seq data (typically >90% zeros) enables specialized storage formats that dramatically reduce memory requirements.
Protocol: Sparse Matrix Implementation
- Assessment: Calculate sparsity ratio (percentage of zeros) in count matrix
- Conversion: Transform dense matrix to sparse representation
- Coordinate Format (COO): Stores (row, column, value) triplets for non-zero elements
- Compressed Sparse Column (CSC): Column-oriented format efficient for column operations
- Memory Monitoring: Compare memory usage before and after conversion
- Compatibility Check: Ensure downstream tools support sparse matrix operations
Implementation in Python:
Algorithm Selection and Parameter Optimization
Computational performance varies significantly across algorithms designed for similar analytical tasks. Informed algorithm selection can reduce computation time from days to hours for large datasets.
Table 2: Computational Characteristics of Common scRNA-seq Algorithms
| Analytical Task | Algorithm | Computational Complexity | Recommended Use Case |
|---|---|---|---|
| Dimensionality Reduction | PCA | O(n³) for exact implementation | Medium-sized datasets (<50,000 cells) |
| Incremental PCA | O(n²) for memory efficiency | Large datasets with memory constraints | |
| UMAP | O(n¹.¹⁴) for approximate implementation | Visualization of large datasets | |
| Clustering | Leiden | O(n log n) for graph traversal | Standard for large single-cell datasets |
| K-means | O(nkdi) for n cells, k clusters, d dimensions, i iterations | Pre-defined cluster number scenarios | |
| DESC | O(nd²) for deep embedding | Batch-corrected clustering | |
| Integration | Harmony | O(nkdi) for n cells, k clusters, d dimensions, i iterations | Fast integration of multiple datasets |
| scVI | O(nd²) for neural network training | Complex batch effects and large datasets |
Protocol: Clustering Parameter Optimization Using Intrinsic Metrics
- Parameter Space Definition: Identify key parameters (resolution, number of neighbors, PCA dimensions)
- Grid Search Setup: Define ranges for each parameter based on dataset size
- Clustering Execution: Run clustering algorithm with each parameter combination
- Intrinsic Metric Calculation: Compute metrics that evaluate cluster quality without ground truth:
- Within-cluster dispersion (lower values indicate compact clusters)
- Banfield-Raftery index (higher values indicate better separation)
- Optimal Parameter Selection: Identify parameter sets that optimize intrinsic metrics
- Biological Validation: Verify that computationally optimal parameters yield biologically meaningful clusters [89]
Research indicates that using UMAP for neighborhood graph generation combined with higher resolution parameters significantly improves clustering accuracy, particularly when using fewer nearest neighbors, which creates sparser graphs that better preserve fine-grained cellular relationships [89].
Integrated Analysis Platforms
For researchers without specialized computational expertise, integrated platforms provide optimized workflows that implement performance best practices automatically.
BestopCloud represents a comprehensive solution that seamlessly integrates multiple analytical modules while managing computational resources efficiently [90]. The platform's modular design allows researchers to execute specific analytical steps independently, then connect results through flexible data flows, preventing unnecessary recomputation.
Protocol: Large-Scale Analysis Using BestopCloud
- Data Upload: Upload raw count matrix in supported formats (H5AD, MTX, or CSV)
- Quality Control Module:
- Set thresholds for mitochondrial gene percentage (<15%) and minimum detected genes (>500)
- Execute filtering to remove low-quality cells
- Integration Module:
- Select batch variables (e.g., patient, condition)
- Apply Harmony integration with default parameters
- Cell Type Annotation:
- Start with SingleR for broad classification
- Refine with ScType using tissue-specific marker genes
- Downstream Analysis: Proceed to differential expression, cell-cell communication, or copy number variation analysis [90]
Performance benchmarks demonstrate that BestopCloud processes a 3,000-cell dataset in approximately 26 seconds, scaling to approximately 3.5 minutes for 20,000 cells in the data processing module [90].
Visualization of Optimized Analytical Workflows
The following diagram illustrates a computationally optimized end-to-end workflow for large-scale scRNA-seq data analysis, incorporating performance-enhancing strategies at each step:
Figure 1: Optimized Computational Workflow for Large-Scale scRNA-seq Data Analysis. This workflow integrates performance-enhancing strategies at each analytical stage, balancing computational efficiency with biological accuracy. Diamond-shaped nodes indicate key optimization points.
Table 3: Research Reagent Solutions for Computational scRNA-seq Analysis
| Resource Category | Specific Tools | Function | Performance Considerations |
|---|---|---|---|
| Programming Environments | R (4.0+), Python (3.8+) | Statistical computing and analysis | R benefits from optimized Bioconductor packages; Python offers better scalability for very large datasets |
| Analysis Packages | Seurat, Scanpy, SingleCellExperiment | Core data structures and analytical methods | Scanpy generally shows better memory efficiency for very large datasets (>50,000 cells) |
| Integration Tools | Harmony, scVI, BBKNN | Batch correction and data integration | Harmony offers fastest computation; scVI provides superior accuracy for complex batch effects |
| Clustering Methods | Leiden, DESC, SC3 | Cell population identification | Leiden is fastest for standard analyses; DESC provides enhanced accuracy with batch correction |
| Visualization Platforms | BestopCloud, CellSnake, UCSC Cell Browser | Interactive exploration and analysis | BestopCloud provides comprehensive functionality; specialized browsers offer domain-specific optimizations |
| Reference Databases | CellTypist, Human Cell Atlas, PanglaoDB | Cell type annotation references | CellTypist offers comprehensive immune cell references; HCA provides broad tissue coverage |
Optimizing computational performance for large-scale scRNA-seq data analysis requires a multifaceted approach that addresses memory utilization, processing efficiency, and algorithmic selection. The protocols and strategies presented in this application note provide a roadmap for researchers to overcome computational bottlenecks while maintaining analytical rigor. As single-cell technologies continue to evolve, producing ever-larger datasets, these optimization approaches will become increasingly essential for extracting biologically meaningful insights in a computationally feasible framework. By implementing feature selection, efficient data structures, parameter optimization, and integrated platforms, researchers can significantly enhance their analytical capabilities while managing computational resources effectively.
Best Practices for Parameter Selection and Algorithm Tuning
Single-cell RNA sequencing (scRNA-seq) has revolutionized the study of cellular heterogeneity by enabling high-resolution analysis of gene expression profiles at the individual cell level [91] [46]. The analytical pipelines for processing scRNA-seq data involve multiple complex steps, each with tunable parameters that significantly impact the reliability and interpretability of results. Clustering inconsistency represents a fundamental challenge, as stochastic processes in clustering algorithms can yield substantially different results across runs, potentially undermining the reliability of assigned cell labels [91]. Similarly, dimensionality reduction methods exhibit varying sensitivity to parameter settings, with some complex models capable of superior performance only after careful tuning [92]. The high-dimensional and sparse nature of scRNA-seq data, compounded by technical artifacts like dropout events, further necessitates optimized parameter selection to distinguish genuine biological signals from noise [93]. This protocol outlines comprehensive strategies for parameter selection and algorithm tuning to enhance the reliability, efficiency, and biological relevance of scRNA-seq analyses.
Theoretical Foundations of scRNA-Seq Analysis
Key Computational Challenges
scRNA-seq data analysis presents several unique computational challenges that parameter tuning aims to address. The "dropout" phenomenon, where genes with actual expression fail to be detected, creates false zeros that distort true gene expression distributions and complicate biological interpretation [93]. The curse of dimensionality arises from measuring thousands of genes across thousands of cells, requiring effective dimensionality reduction to visualize and analyze cellular relationships. Technical variability between experiments, protocols, and sequencing batches introduces noise that can obscure biological signals without proper normalization and batch correction [46] [92]. Additionally, algorithmic stochasticity in methods like Leiden clustering can produce different results across runs with different random seeds, potentially leading to inconsistent cell type identification [91].
Critical Trade-offs in Parameter Selection
Parameter optimization in scRNA-seq analysis involves balancing several competing analytical priorities. Resolution versus robustness represents a fundamental trade-off, where higher clustering resolution parameters may identify finer cell subtypes but with increased vulnerability to noise and reduced consistency across runs [91]. The complexity versus interpretability balance pits sophisticated models like variational autoencoders against simpler PCA-based approaches, with the former potentially capturing more nuanced patterns but requiring extensive tuning and offering less transparent mechanics [92] [94]. Computational efficiency versus analytical depth must be considered, as more comprehensive consistency evaluations and parameter sweeps produce more reliable results but require substantially greater computational resources [91] [92].
Quantitative Benchmarking of Method Performance
Performance Metrics for Algorithm Evaluation
Systematic evaluation of analytical methods requires standardized metrics that quantify performance across diverse datasets. The inconsistency coefficient (IC) measures clustering stability across multiple runs with different random seeds, with values approaching 1 indicating highly consistent results [91]. Element-centric similarity (ECS) quantifies the agreement between different cluster labels by comparing affinity matrices derived from cluster memberships, providing an unbiased similarity assessment [91]. The silhouette coefficient evaluates clustering quality by measuring how similar cells are to their own cluster compared to other clusters, with higher values indicating better-defined clusters [92] [93]. Adjusted mutual information (AMI) assesses how well computational clustering recovers known cell type annotations, serving as a proxy for practical utility in cell type identification [92].
Table 1: Performance Metrics for scRNA-Seq Algorithm Evaluation
| Metric | Calculation | Interpretation | Optimal Range |
|---|---|---|---|
| Inconsistency Coefficient (IC) | Inverse of pSpT where S is similarity matrix, p is probability vector | Measures clustering stability across runs | Closer to 1 indicates higher consistency [91] |
| Element-Centric Similarity (ECS) | Average of ECS vector derived from affinity matrix differences | Quantifies agreement between clustering results | 0-1, higher values indicate better agreement [91] |
| Silhouette Coefficient | (b - a)/max(a,b) where a=intra-cluster, b=inter-cluster distance | Measures clustering compactness and separation | -1 to 1, higher values better [93] |
| Adjusted Mutual Information (AMI) | Mutual information adjusted for chance agreement | Measures cell type identification accuracy | 0-1, higher values better [92] |
Benchmarking Results Across Method Categories
Empirical benchmarking reveals how different categories of scRNA-seq analysis methods perform under varying parameter configurations. Dimensionality reduction methods show distinct performance characteristics, with PCA-based approaches like scran and Seurat performing competitively with default parameters but benefiting minimally from tuning, while more complex models like ZinbWave, DCA, and scVI can achieve superior performance but only after extensive parameter optimization [92]. Clustering consistency methods demonstrate substantial speed variation, with the recently developed scICE framework achieving up to 30-fold improvement in speed compared to conventional consensus clustering approaches like multiK and chooseR while maintaining accuracy [91]. Cell type annotation tools show that simpler statistical approaches like PCLDA (combining PCA and linear discriminant analysis) can achieve performance comparable to or better than more complex machine learning methods, particularly when reference and query data come from different protocols [94].
Table 2: Benchmarking Results of scRNA-Seq Analysis Methods
| Method Category | Representative Tools | Performance with Default Parameters | Performance After Tuning | Computational Efficiency |
|---|---|---|---|---|
| Dimensionality Reduction | scran, Seurat | Competitive (AMI: 0.75-0.84) [92] | Minimal improvement [92] | High [92] |
| Dimensionality Reduction | ZinbWave, DCA, scVI | Variable (AMI: 0.56-0.79) [92] | Substantial improvement possible [92] | Medium to Low (requires tuning) [92] |
| Clustering Consistency | multiK, chooseR | Requires consensus matrix construction [91] | Limited by computational cost [91] | Low (high computational cost) [91] |
| Clustering Consistency | scICE | Identifies consistent clustering results [91] | Evaluates consistency across resolutions [91] | High (30x faster than alternatives) [91] |
| Cell Type Annotation | Complex machine learning | Variable across protocols [94] | Requires retraining for new protocols [94] | Medium to Low [94] |
| Cell Type Annotation | PCLDA (PCA + LDA) | Stable across protocols [94] | Minimal tuning required [94] | High [94] |
Experimental Protocols for Parameter Optimization
Protocol 1: Clustering Consistency Evaluation with scICE
Purpose: To evaluate clustering consistency across multiple runs and identify reliable cluster labels while minimizing computational burden.
Principles: Traditional clustering evaluation requires constructing computationally expensive consensus matrices, but scICE uses the inconsistency coefficient (IC) and parallel processing to achieve up to 30-fold speed improvement [91]. The protocol assesses label consistency across multiple clustering runs with different random seeds, systematically identifying consistent clustering results.
Materials:
- Quality-controlled scRNA-seq count matrix
- High-performance computing environment with multiple cores
- scICE software (available as R/Python package)
Procedure:
- Data Preprocessing: Perform standard quality control to filter low-quality cells and genes. Apply dimensionality reduction (e.g., with scLENS) to reduce data size while preserving biological signals [91].
- Graph Construction: Build a cell-cell graph based on distances in the reduced dimensionality space.
- Parallel Clustering: Distribute the graph to multiple processes across computing cores. Run Leiden clustering simultaneously on each process with different random seeds [91].
- Similarity Calculation: For each pair of cluster labels, compute element-centric similarity (ECS) by:
- Calculating difference in affinity matrices between label pairs
- Summing row-wise to obtain L1 vector representing total affinity difference per cell
- Subtracting L1 vector from 1 to obtain ECS vector indicating membership agreement [91]
- Inconsistency Coefficient Calculation: Construct similarity matrix S where elements Sij represent similarity between labels ci and cj. Calculate IC as the inverse of pSpT where p is the probability vector of different label occurrences [91].
- Interpretation: IC values close to 1 indicate high consistency, while values progressively higher than 1 indicate increasing inconsistency. Identify clustering resolutions with IC ≈ 1 for reliable downstream analysis.
Troubleshooting:
- If IC values are consistently >1.05 across resolutions, consider increasing the number of parallel clustering runs (default: 50-100 runs).
- If computational resources are limited, reduce the number of clustering runs, but note that this may reduce consistency assessment reliability.
- For extremely large datasets (>50,000 cells), consider initial subsetting to estimate appropriate resolution parameters before full analysis.
Protocol 2: Dimensionality Reduction Parameter Tuning
Purpose: To systematically optimize parameters for dimensionality reduction methods to maximize cell type separation and downstream analysis performance.
Principles: Dimensionality reduction is a critical first step for many scRNA-seq analyses including visualization, clustering, and trajectory inference. Performance varies significantly across methods and parameter settings, with complex models being particularly sensitive to tuning [92].
Materials:
- Normalized scRNA-seq count matrix
- Benchmark datasets with known cell type annotations (for validation)
- Computing environment with adequate resources for parameter sweeps
Procedure:
- Method Selection: Choose representative methods from different algorithmic families:
- Parameter Space Definition: Identify tunable parameters for each method:
- PCA-based: Number of principal components, variable gene selection threshold
- ZinbWave: Number of factors, regularization parameters
- DCA: Network architecture, dropout rate, number of epochs
- scVI: Number of hidden layers, learning rate, number of epochs [92]
- Performance Evaluation: For each parameter combination, run dimensionality reduction and evaluate using:
- Silhouette coefficient on known cell types
- Adjusted mutual information (AMI) of k-means clusters versus known cell types [92]
- Optimal Parameter Selection: Identify parameter sets that maximize both silhouette and AMI scores. Prioritize parameters that perform consistently well across multiple benchmark datasets.
- Validation: Apply optimized parameters to independent validation datasets to assess generalizability.
Troubleshooting:
- If performance metrics show high variance across datasets, increase the number of benchmark datasets used for tuning.
- If computational requirements become prohibitive, implement Bayesian optimization or random search instead of exhaustive grid search.
- For methods like scVI that require GPU acceleration, ensure compatible hardware and software environments.
Protocol 3: Imputation Method Optimization with scVGAMF
Purpose: To optimize imputation of dropout events in scRNA-seq data by integrating both linear and non-linear features.
Principles: scVGAMF addresses dropout imputation by combining non-negative matrix factorization (NMF) for linear features and variational graph autoencoders (VGAE) for non-linear features, outperforming methods that rely exclusively on one approach [93].
Materials:
- Raw scRNA-seq count matrix
- Computational environment with Python and deep learning capabilities
- scVGAMF software implementation
Procedure:
- Data Preparation:
- Perform logarithmic normalization of raw count matrix
- Identify highly variable genes using variance stabilizing transformation
- Partition genes into groups (default: 2000 genes per group) [93]
- Cell Clustering:
- Apply PCA to each gene group
- Perform spectral clustering with cluster numbers ranging from 4 to 15
- Select optimal cluster number using silhouette coefficient [93]
- Similarity Matrix Calculation:
- Compute cell similarity matrix by integrating Pearson correlation, Spearman correlation, and Cosine similarity using geometric mean
- Compute gene similarity matrix using Jaccard index based on co-expression patterns [93]
- Feature Integration:
- Apply VGAE to cell and gene similarity matrices to extract non-linear features
- Apply NMF to gene expression submatrices to extract linear features
- Integrate both feature types using fully connected neural network for final imputation [93]
- Validation:
- Evaluate imputation accuracy on simulated dropout datasets
- Assess downstream analysis improvements in cell clustering, differential expression, and trajectory inference
Troubleshooting:
- If imputation introduces excessive noise, adjust the balance between linear and non-linear components.
- If computational requirements are too high, reduce gene group size or implement mini-batch processing.
- Validate imputation results using positive control genes with known expression patterns.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Essential Research Reagents and Computational Solutions for scRNA-Seq Analysis
| Category | Item | Function/Purpose | Example Tools/Protocols |
|---|---|---|---|
| Wet Lab Protocols | Drop-seq [46] | High-throughput, low-cost 3' end counting | Droplet-based cell isolation [46] |
| Wet Lab Protocols | Smart-Seq2 [46] | Full-length transcript sequencing with enhanced sensitivity | FACS-based cell isolation [46] |
| Wet Lab Protocols | snRNA-seq [46] | Single-nucleus RNA sequencing for fragile or frozen samples | Nuclei isolation [46] |
| Wet Lab Protocols | Cell Preparation Handbook [95] | Best practices for viable single-cell suspension preparation | 10x Genomics protocols [95] |
| Computational Tools | scICE [91] | Clustering consistency evaluation with parallel processing | Leiden clustering consistency [91] |
| Computational Tools | scVGAMF [93] | Dropout imputation integrating linear and non-linear features | VGAE + NMF integration [93] |
| Computational Tools | PCLDA [94] | Interpretable cell type annotation using PCA and LDA | t-test gene screening + PCA + LDA [94] |
| Computational Tools | Seurat [92] | PCA-based dimensionality reduction and analysis | scRNA-seq preprocessing and clustering [92] |
| Computational Tools | scVI [92] | Variational autoencoder for dimensionality reduction | Deep learning-based representation [92] |
Implementation Strategies and Practical Guidelines
Developing an Optimization Framework
Effective parameter tuning requires a systematic framework that balances computational efficiency with analytical rigor. Stratified tuning approaches prioritize tuning effort based on method sensitivity, with complex models like scVI requiring extensive optimization while simpler methods like PCA-based dimensionality reduction perform well with defaults or minimal tuning [92]. Multi-resolution consistency checking implements scICE across a range of clustering resolutions to identify consistently stable clustering solutions, substantially narrowing the candidate cluster numbers that require further biological validation [91]. Benchmark-driven validation uses datasets with known cell type composition to establish performance baselines and identify parameter sets that generalize well across diverse biological contexts [92].
Interpretation and Quality Assessment
Robust interpretation of tuning results requires careful quality assessment beyond simple metric optimization. Consistency-reliability alignment ensures that computationally consistent results align with biological expectations, using marker gene expression and known cell type signatures to validate parameter choices [91]. Dropout pattern awareness recognizes that optimal imputation parameters must distinguish technical zeros (dropouts) from biological zeros (genuine absence of expression), with methods like scVGAMF implementing specific clustering-based approaches to maintain this distinction [93]. Interpretability-complexity balancing favors approaches that provide transparent decision boundaries when possible, as demonstrated by PCLDA's use of linear discriminant analysis after feature selection, enabling clear biological interpretation of cell type classification decisions [94].
Systematic parameter selection and algorithm tuning are essential components of robust scRNA-seq analysis. The protocols presented here for clustering consistency evaluation, dimensionality reduction optimization, and dropout imputation provide structured approaches to enhance analytical reliability. The key insight across these domains is that method performance varies significantly with parameter settings, with complex models offering potential performance advantages but requiring more extensive tuning. Computational efficiency remains a practical constraint, with newer methods like scICE demonstrating substantial improvements in speed without sacrificing reliability. Interpretability continues to be an important consideration, with simpler statistical approaches often performing comparably to black-box machine learning methods while offering greater transparency. By implementing these best practices for parameter selection and algorithm tuning, researchers can enhance the reliability, efficiency, and biological relevance of their scRNA-seq analyses, leading to more robust discoveries in cellular heterogeneity and function.
Ensuring Robust Biological Insights: Validation, Benchmarking, and Novel Cell Type Discovery
Within the broader thesis on single-cell RNA sequencing (scRNA-seq) data analysis, this application note addresses a critical methodological challenge: the validation of computational findings. Single-cell technologies provide unprecedented resolution for dissecting cellular heterogeneity, inferring cell-cell communication, and identifying novel cell states or markers [96]. However, the predominantly descriptive nature of scRNA-seq studies necessitates robust validation frameworks to translate observations into biologically meaningful and therapeutically relevant insights [97]. A central challenge remains the functional validation of hypothesized cell-cell interactions and marker genes, which often generate lengthy lists of candidates that are impossible to probe experimentally in their entirety [96].
This protocol details a structured framework that leverages bulk RNA-seq data and public atlases to prioritize and validate scRNA-seq-derived findings. By integrating computational cross-referencing with experimental confirmation, we provide a systematic pathway from target identification to functional assessment, thereby bridging the gap between large-scale genomic data and biological insight. This approach is designed to enhance the reliability of conclusions drawn from single-cell studies and is particularly vital for researchers and drug development professionals aiming to identify and characterize novel therapeutic targets.
Conceptual Framework for scRNA-seq Validation
The validation of scRNA-seq data operates on multiple levels, from confirming the presence and identity of cell populations to establishing the functional role of specific genes. A multi-faceted approach is essential for building confidence in single-cell findings.
- Spatial Confirmation: Techniques like RNA Fluorescence In Situ Hybridization (RNA FISH) and Immunofluorescence (IF) are used to validate the spatial localization of identified cell types or marker genes within the native tissue architecture, confirming that computationally identified populations have a physical correlate [98].
- Protein-level Verification: Methods such as Immunohistochemistry (IHC) and flow cytometry confirm that transcriptomic signatures translate to the protein level, a critical consideration for drug target development [98].
- Functional Validation: Gene overexpression, silencing (e.g., RNAi), and knockout (e.g., CRISPR/Cas9) experiments in relevant in vitro or in vivo models are the ultimate test for the functional role of a candidate gene identified through differential expression analysis [98] [97].
- Multi-omics Integration: Correlating scRNA-seq data with other data modalities, such as single-cell ATAC-seq (assaying chromatin accessibility) or spatial transcriptomics, provides orthogonal validation and a more comprehensive understanding of regulatory mechanisms [98].
Bulk RNA-seq data and large public atlases serve as powerful, readily accessible resources for the initial cross-referencing and prioritization of candidates before embarking on more resource-intensive experimental validations. This framework is exemplified in a study on tip endothelial cells, where researchers used a rigorous in silico prioritization workflow, incorporating criteria from the Guidelines On Target Assessment for Innovative Therapeutics (GOT-IT), to select six candidate genes from a list of over 50 top-ranking markers for subsequent functional validation [97].
Computational Cross-Referencing with Bulk RNA-seq
Rationale and Workflow
Bulk RNA-seq remains a widely available and cost-effective technology, especially in clinical settings. Deconvolution methods allow researchers to extract cell-type-specific signals from bulk RNA-seq data using signatures derived from scRNA-seq. This enables the validation of scRNA-seq findings in larger, independent cohorts profiled with bulk RNA-seq. The pathway-level information extractor (PLIER) algorithm can be adapted to learn a single-cell-informed deconvolution specific to a given biological context, producing interpretable latent variables for analysis of bulk data [99].
The following diagram illustrates the core workflow for leveraging bulk RNA-seq to validate findings from a single-cell study:
Protocol: Using CLIER for Bulk RNA-seq Validation
This protocol details the steps for using the CLIER (PLIER model trained on single-cell signatures) approach to validate scRNA-seq findings with bulk data [100] [99].
Step 1: Construct a Single-Cell Signature Atlas
- Collect scRNA-seq data from your study and relevant public datasets.
- Identify and annotate cell populations (e.g., clustering, marker genes).
- Define gene expression signatures for each population or state of interest. These can be marker genes, differentially expressed genes, or pathway scores.
Step 2: Train a CLIER Model
- Input the single-cell-derived gene signatures into the PLIER algorithm to train a CLIER model.
- The model learns a set of latent variables (LVs), each representing a biological pathway or cell-type-specific signature derived from the single-cell data.
Step 3: Apply the Model to a Bulk RNA-seq Dataset
- Obtain a bulk RNA-seq dataset from an independent cohort relevant to your research question (e.g., from GEO, TCGA, or GTEx) [101].
- Use the trained CLIER model to transform the bulk dataset. This step generates a matrix of LV scores for each sample in the bulk dataset.
Step 4: Perform Validation Analyses
- Correlate LV scores with clinical or phenotypic metadata (e.g., disease severity, treatment response) to confirm the biological relevance of the single-cell-identified populations.
- Test if the LV corresponding to your cell type of interest is significantly associated with the expected condition in the bulk data.
Key Public Bulk RNA-seq Databases
The table below summarizes essential public repositories for sourcing bulk RNA-seq data for validation purposes [101].
Table 1: Key Public Databases for Bulk RNA-Seq Data
| Database Name | Description | Key Features | Data Access |
|---|---|---|---|
| GEO (Gene Expression Omnibus) [101] | NIH-hosted repository for functional genomics data. | Includes diverse platforms and organisms; links to SRA for raw data (FASTQ). | Count matrices via accession page; FASTQ via SRA. |
| EMBL Expression Atlas [101] | EMBL's resource for curated RNA-seq datasets. | Datasets categorized as "baseline" or "differential"; enhanced browsing. | Processed data via "Downloads" tab. |
| GTEx (Genotype-Tissue Expression) [101] | Resource for normal human tissue expression. | Bulk and single-nucleus RNA-seq; QTL data; tissue-specific exploration. | Data downloadable by tissue type. |
| TCGA (The Cancer Genome Atlas) [101] | NIH repository for cancer genomics. | Linked to GDC portal; rich clinical and molecular data. | RNA-seq counts per sample; requires file combination. |
| Recount3 [101] | Uniformly processed resource for GEO/SRA, GTEx, TCGA. | Normalized data accessible via R/Bioconductor. | recount3 R package for data retrieval. |
Leveraging Large-Scale Public Single-Cell Atlases
The Role of Consolidated Atlases
Recent initiatives have created massive, consolidated single-cell atlases that serve as invaluable benchmarks for validation. These resources provide a comprehensive baseline of cell states across tissues, species, and conditions, allowing researchers to contextualize their own findings and assess the generalizability of identified cell types or gene signatures.
The recent launch of the Arc Virtual Cell Atlas is a prime example, combining data from over 300 million cells [100] [102]. Its inaugural release includes two key datasets:
- Tahoe-100M: The world's largest single-cell perturbation dataset, with 100 million cells and 60,000 drug-cell interactions across 50 cancer cell lines [100] [102].
- scBaseCount: A curated repository of public scRNA-seq data from 200 million cells across 21 species, standardized using AI agents to ensure interoperability for machine learning [100] [102].
Protocol: Atlas-Based Contextualization and Validation
The logical flow for using a public atlas for validation involves querying, comparing, and interpreting results against a known reference.
Step 1: Identify a Candidate from Your scRNA-seq Data
- This could be a novel cell state, a candidate biomarker gene, or a hypothesized ligand-receptor pair inferred from cell-cell communication analysis.
Step 2: Query the Public Atlas
Step 3: Perform Contextual Validation
- Conservation Check: Assess if the marker gene is expressed in analogous cell types across multiple species (if using a multi-species atlas like scBaseCount).
- Specificity Check: Determine whether the candidate is specific to your biological context (e.g., disease) or is a general feature of the tissue.
- Perturbation Response: Using a resource like Tahoe-100M, investigate how your candidate gene or cell state responds to drug treatments or other perturbations, which can support its role as a therapeutic target [100].
Key Public Single-Cell Databases
The table below lists major public databases specifically for scRNA-seq data that are essential for validation and contextualization [101].
Table 2: Key Public Databases for Single-Cell RNA-Seq Data
| Database Name | Description | Key Features | Data Access |
|---|---|---|---|
| Single Cell Portal (Broad) [101] | Broad Institute's portal for scRNA-seq studies. | Search by organ, species, disease; built-in visualizations (UMAP). | Raw/normalized data after login. |
| CZ Cell x Gene Discover [101] | Chan Zuckerberg Initiative's database. | Hosts 500+ datasets; exploration via cellxgene tool. | Easy download of datasets. |
| PanglaoDB [101] | Karolinska Institutet's database. | Access to 1300+ experiments; exploration of markers. | R objects or text files. |
| scRNAseq (Bioconductor) [101] | R/Bioconductor package with curated datasets. | Datasets as SingleCellExperiment objects; easy downstream analysis. | Direct download via R code. |
| Allen Brain Cell Atlas [101] | Survey of single-cell data in mouse and human brain. | Hierarchical exploration of brain cell types. | Online exploration and download. |
A Practical Guide to Gene Prioritization and Validation
Translating scRNA-seq candidate lists into validated targets requires a systematic prioritization funnel. The following workflow, adapted from a study on tip endothelial cells, integrates cross-referencing with bulk data and public atlases to select the most promising candidates for functional assays [97].
Phase 1: In Silico Prioritization
- Input: A ranked list of marker genes from differential expression or network analysis.
- Criterion 1: Target-Disease Linkage: Justify the biological context. In the tip cell study, candidates were restricted to those enriched in tumor-derived tip ECs, as this phenotype is clinically relevant and sensitive to therapy [97].
- Criterion 2: Target-Related Safety: Exclude genes with known genetic links to other diseases (e.g., in the central nervous system) based on literature review [97].
- Criterion 3: Strategic Novelty: Focus on genes minimally described in the context of your study (e.g., angiogenesis). This helps identify novel targets and avoids crowded intellectual property space [97].
- Criterion 4: Technical Feasibility: Filter for genes with available perturbation tools (e.g., siRNAs), non-secreted proteins for easier targeting, and specific expression in your cell type of interest compared to all other cell types in a public tissue atlas (e.g., log-fold change >1) [97].
Phase 2: Experimental Functional Validation
- In Vitro Models: Use relevant primary cells or cell lines.
- Efficient Knockdown: Employ multiple siRNAs per gene to ensure robust knockdown and control for off-target effects [97].
- Phenotypic Assays: Test the functional role of candidates using assays relevant to the cell type.
The Scientist's Toolkit
The table below outlines key reagents and computational tools essential for executing the validation protocols described in this document.
Table 3: Research Reagent and Tool Solutions for scRNA-seq Validation
| Item/Tool | Function/Description | Example Use Case |
|---|---|---|
| Parse Biosciences' GigaLab [100] | Single-cell RNA sequencing platform for scalable sample preparation. | Generation of large-scale validation datasets (e.g., Tahoe-100M). |
| siRNA Oligos [97] | Synthetic small interfering RNAs for transient gene knockdown. | Functional validation of candidate genes in in vitro models (HUVECs). |
| RNeasy Mini Kit (QIAGEN) [103] | Isolation of high-quality total RNA from cell cultures. | RNA preparation for bulk RNA-seq or qPCR validation. |
| DESeq2 [103] | R/Bioconductor package for differential expression analysis of bulk or single-cell count data. | Identifying differentially expressed genes in validation cohorts. |
| CellPhoneDB [96] | Open-source tool for inferring cell-cell communication from scRNA-seq data. | Validation of hypothesized ligand-receptor interactions. |
| CLIER/PLIER Algorithm [99] | Computational method for extracting latent variables from bulk data using single-cell signatures. | Deconvolution and cross-referencing of bulk RNA-seq datasets. |
Benchmarking Computational Tools for Performance and Accuracy
Within the broader context of single-cell RNA sequencing (scRNA-seq) data analysis research, the selection of appropriate computational tools is as critical as the choice of experimental protocols. The performance of these tools directly impacts the biological interpretation of data, influencing downstream conclusions in research and drug development. As the field moves toward constructing comprehensive cell atlases and refining clinical diagnostics, the need for rigorous, independent benchmarking of computational methods has become paramount [104]. This application note synthesizes recent benchmarking studies to provide validated protocols and evidence-based recommendations for computational tool selection, ensuring researchers and scientists can achieve accurate and reproducible results.
Quantitative Benchmarking of scRNA-seq Tools
Benchmarking Single-Cell Clustering Algorithms
Clustering is a foundational step in scRNA-seq analysis for identifying cell types and states. A comprehensive 2025 benchmark evaluated 28 clustering algorithms on 10 paired transcriptomic and proteomic datasets, assessing performance using Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), computational resource use, and robustness [105].
Table 1: Top-Performing Single-Cell Clustering Algorithms (2025 Benchmark)
| Method | Overall Rank (Transcriptomics) | Overall Rank (Proteomics) | Key Strength | Recommended Use Case |
|---|---|---|---|---|
| scAIDE | 2 | 1 | Top performance in proteomics | Studies prioritizing protein data analysis |
| scDCC | 1 | 2 | Excellent balance of performance & memory efficiency | Large-scale studies with limited computational resources |
| FlowSOM | 3 | 3 | High robustness & strong cross-omics performance | General-purpose use & multi-omics integration |
| TSCAN | 6 | 4 | High time efficiency | Studies requiring rapid analysis turnaround |
| SHARP | 7 | 5 | High time efficiency | Large dataset analysis with time constraints |
The study revealed that top-performing methods like scDCC, scAIDE, and FlowSOM demonstrated consistent high performance across both transcriptomic and proteomic modalities, indicating strong generalizability [105]. In contrast, some methods like CarDEC and PARC showed significant performance disparity between modalities, ranking highly in transcriptomics but dropping substantially in proteomics. This highlights the importance of selecting tools validated for specific data modalities.
Benchmarking Copy Number Variation Callers
In cancer genomics, accurately inferring copy number variations (CNVs) from scRNA-seq data is crucial for understanding tumor evolution and heterogeneity. A 2025 benchmarking study evaluated six popular CNV callers across 21 scRNA-seq datasets, comparing results to orthogonal ground truth measurements from (sc)WGS or WES [106].
Table 2: Performance Characteristics of scRNA-seq CNV Inference Tools
| Method | Underlying Data | Output Resolution | Key Strength | Performance Note |
|---|---|---|---|---|
| CaSpER | Expression + Allelic Information | Per cell | Robust performance in droplet-based data | Balanced CNV inference, accurate calls in clinical samples |
| CopyKAT | Expression only | Per cell | Excellent tumor subpopulation identification | Consistent, balanced performance |
| InferCNV | Expression only | Subclones | High sensitivity for tumor subclones | Excels in single-platform studies |
| Numbat | Expression + Allelic Information | Subclones | Robustness to batch effects | Better for large droplet datasets but high runtime |
| SCEVAN | Expression only | Subclones | Good segmentation approach | Platform-dependent performance |
| CONICSmat | Expression only | Chromosome arm | Per-chromosome arm resolution | Lower sensitivity |
The evaluation found that methods incorporating allelic information (e.g., CaSpER, Numbat) generally performed more robustly for large droplet-based datasets, though they required higher computational runtime [106]. For research focused on identifying tumor subpopulations, inferCNV and CopyKAT were particularly effective. Batch effects significantly impacted most methods when integrating datasets from different platforms, necessitating the use of batch correction tools like ComBat.
Experimental Protocols for Benchmarking Studies
Standardized Workflow for scRNA-seq CNV Caller Evaluation
Objective: To rigorously evaluate the performance of CNV calling methods on scRNA-seq data using orthogonal validation [106].
Input Data Requirements:
- scRNA-seq datasets from both droplet-based (e.g., 10x Genomics) and plate-based (e.g., SMART-seq) platforms
- Matched ground truth CNV data from (sc)WGS, WES, or cell lines with known CNV profiles
- Reference diploid cells (either from the same sample or matched external datasets)
Procedure:
- Data Preparation and Preprocessing
- Obtain scRNA-seq count matrices and corresponding ground truth CNV profiles
- For primary tissues, manually annotate healthy (diploid) cells to use as reference
- For cancer cell lines, select matched external reference datasets from similar cell types
Method Application
- Run each CNV caller (e.g., CaSpER, inferCNV, CopyKAT) according to developer specifications
- Use the same reference cells for all methods to ensure comparability
- Generate both discrete CNV calls and normalized expression scores where available
Performance Assessment
- Calculate pseudobulk CNV profiles by averaging per-cell results
- Compare to ground truth using correlation, AUC scores, and partial AUC
- Determine sensitivity and specificity for gain/loss detection using biologically meaningful thresholds
- Evaluate computational requirements (runtime and memory)
Downstream Analysis Validation
- Assess accuracy in identifying tumor subpopulations
- Test robustness to batch effects across platforms
- Validate findings on clinical samples with orthogonal genomic data
Expected Outputs:
- Quantitative performance metrics (correlation, AUC, F1 scores) for each method
- Assessment of method strengths/limitations under different experimental conditions
- Recommendations for optimal tool selection based on data characteristics
CNV Caller Benchmarking Workflow
Protocol for Cross-Modal Clustering Algorithm Assessment
Objective: To evaluate clustering algorithm performance across single-cell transcriptomic and proteomic data modalities [105].
Input Data Requirements:
- Paired single-cell transcriptomic and proteomic datasets (e.g., from CITE-seq, ECCITE-seq)
- Curated cell type annotations at different granularity levels
- 30 simulated datasets with varying noise levels and dataset sizes
Procedure:
- Data Acquisition and Curation
- Obtain 10+ real datasets from sources like SPDB (Single-Cell Proteomic DataBase)
- Include datasets with cell type labels at different resolution levels
- Generate simulated datasets with controlled noise parameters
Algorithm Configuration
- Select 28 clustering algorithms spanning machine learning, community detection, and deep learning approaches
- Apply consistent preprocessing and Highly Variable Gene (HVG) selection
- Use default parameters as specified by developers
Performance Evaluation
- Calculate ARI, NMI, Clustering Accuracy, and Purity against ground truth
- Measure peak memory usage and running time for each method
- Assess impact of HVG selection and cell type granularity
- Evaluate robustness using simulated datasets with varying noise
Multi-Omics Integration Analysis
- Apply 7 feature integration methods (e.g., moETM, sciPENN, totalVI)
- Test single-omics clustering algorithms on integrated features
- Compare performance gains from multi-omics integration
Expected Outputs:
- Ranking of clustering methods by modality and performance metric
- Guidance on optimal method selection for specific data types
- Assessment of multi-omics integration benefits for clustering
Table 3: Key Research Reagent Solutions for scRNA-seq Benchmarking
| Resource Type | Specific Examples | Function/Purpose | Considerations |
|---|---|---|---|
| scRNA-seq Platforms | 10x Chromium, BD Rhapsody | Generate 3' scRNA-seq data from complex tissues | Cell type detection biases exist between platforms [107] |
| Full-Length Protocols | SMART-seq3, G&T-seq, Takara SMART-seq HT | Sensitive full-length transcriptome profiling | Higher gene detection; essential for splice variants, mutations [108] |
| Reference Datasets | Human PBMCs, Cell line mixtures (e.g., HEK293, NIH3T3) | Standardized samples for method comparison | Enable cross-platform performance assessment [109] [110] |
| Benchmarking Pipelines | Snakemake CNV benchmarking [106] | Reproducible evaluation of new methods | Facilitates method comparison and optimization |
| Data Integration Tools | Harmony [109], Combat | Batch effect correction for cross-platform data | Essential when combining datasets from different technologies |
Integrated Tool Selection Framework
Choosing optimal computational tools requires matching method capabilities to specific research goals and data characteristics. The following decision framework synthesizes findings from multiple benchmarking studies:
Computational Tool Selection Framework
Application Guidelines:
For cell atlas construction and cell type identification: Prioritize scDCC for its balance of performance and memory efficiency, or FlowSOM for its robustness across data modalities [105]. Always validate clustering results with known marker genes.
For cancer genomics and CNV detection: Select CaSpER for droplet-based data when allelic information is available, or CopyKAT for robust tumor subpopulation identification [106]. Use inferCNV for focused analysis on single-platform data.
For studies requiring data integration: Implement batch correction tools like Harmony [109] or ComBat when combining datasets from different platforms, as batch effects significantly impact most CNV detection and clustering methods.
For resource-constrained environments: Consider TSCAN or SHARP for clustering when time efficiency is critical, or scDCC when memory resources are limited [105].
This framework provides an evidence-based starting point for tool selection, though researchers should validate choices using their specific data characteristics when possible.
Comparative Analysis of Datasets with Tools like scCompare and scVI
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling the unbiased assessment of cellular phenotypes at unprecedented resolution, allowing researchers to interrogate gene expression transcriptome-wide in individual cells [111]. This technology has become instrumental in diverse fields including development, autoimmunity, and cancer research, yet the analysis of scRNA-seq data presents significant challenges due to its high-dimensional and sparse nature, technical noise, batch effects, and cellular heterogeneity [112]. A fundamental question in downstream analysis is how to evaluate biological similarities and differences between samples in this high-dimensional space, particularly when dealing with cellular heterogeneity within samples [113] [111].
To address these challenges, computational biologists have developed specialized tools for the comparative analysis of scRNA-seq datasets. Two prominent approaches with distinct methodological foundations are scCompare (Single-cell Compare) and scVI (single-cell Variational Inference). scCompare operates as a computational pipeline that utilizes correlation-based mapping and statistical thresholding to transfer phenotypic identities between datasets while facilitating novel cell type detection [113] [111]. In contrast, scVI employs a deep generative modeling framework based on variational inference to learn a probabilistic representation of gene expression data, enabling multiple downstream analysis tasks through the same unified model [114] [112]. This article provides detailed application notes and protocols for employing these tools in comparative scRNA-seq analyses, with content structured within the broader context of single-cell RNA sequencing data analysis research for an audience of researchers, scientists, and drug development professionals.
Key Characteristics of scCompare and scVI
scCompare is designed specifically for comparing scRNA-seq datasets and mapping phenotypic labels from a reference dataset to a target dataset. Its methodology is based on constructing cell type-specific prototype signatures from averaged gene expression profiles of annotated cell populations in the reference data [111]. The tool then employs statistical thresholding derived from correlation distributions to determine whether cells in the target dataset should be assigned known phenotypic labels or classified as "unmapped" to facilitate novel cell type discovery [111]. This approach provides interpretable results and explicitly handles cellular heterogeneity by allowing for unannotated cell populations.
scVI represents a fundamentally different approach based on probabilistic modeling. It posits a flexible generative model of scRNA-seq count data using a variational autoencoder framework, where observed UMI counts are modeled through a hierarchical Bayesian model with conditional distributions specified by deep neural networks [114] [112]. The model captures gene expression through latent variables representing biological state and technical noise, accounting for batch effects, limited sensitivity, and over-dispersion through a zero-inflated negative binomial distribution [112]. This learned representation then supports multiple downstream tasks including dimensionality reduction, differential expression, batch correction, and transfer learning.
Performance Comparison in Benchmark Studies
A benchmark evaluation using scRNA-seq data from human peripheral blood mononuclear cells (PBMCs) demonstrated that scCompare outperformed scVI in higher precision and sensitivity for most cell types [111] [115]. This performance advantage is attributed to scCompare's focused design for dataset comparison and label transfer tasks, whereas scVI serves as a more general-purpose framework for multiple analysis tasks. However, scVI exhibits significant advantages in scalability, capable of processing datasets exceeding one million cells, and provides a unified probabilistic model consistent across different analysis tasks [112].
Table 1: Performance Characteristics of scCompare and scVI
| Feature | scCompare | scVI |
|---|---|---|
| Primary Function | Dataset comparison and phenotypic label transfer | Probabilistic representation and multi-task analysis |
| Methodological Approach | Correlation-based mapping with statistical thresholding | Deep generative modeling with variational inference |
| Novel Cell Type Detection | Explicitly supported via "unmapped" classification | Indirectly through latent representation |
| Scalability | Suitable for large datasets [111] | Optimized for very large datasets (>1 million cells) [114] [112] |
| Batch Effect Handling | Not explicitly described | Explicit modeling and correction [112] |
| Interpretability | High (direct correlation-based approach) | Lower (black-box neural networks) |
| Benchmark Performance | Higher precision and sensitivity for most cell types [111] | Competitive general-purpose performance |
Experimental Protocols
Protocol for scCompare: Phenotypic Label Transfer and Novelty Detection
Data Preprocessing and Reference Signatures
Begin with standard scRNA-seq preprocessing using Scanpy or Seurat to filter low-quality cells, normalize counts, and identify highly variable genes [111]. For the reference dataset, perform clustering using the Leiden algorithm and annotate cell populations based on established markers or prior knowledge. To generate phenotypic prototype signatures:
- Calculate the average expression of highly variable genes for each annotated cell population.
- For each phenotypic label, compute the distribution of Pearson correlation coefficients between individual cells and their corresponding prototype signature.
- Establish statistical thresholds for phenotype inclusivity using Median Absolute Deviation (MAD). The default cutoff is 5*MAD below the median correlation, though this parameter can be adjusted for more or less stringent mapping [111].
Mapping and Novelty Detection
For the target dataset, process cells through the following workflow:
- Calculate correlation coefficients between each cell's expression profile and all reference phenotypic prototypes.
- Assign each cell to the phenotypic label with the highest correlation coefficient.
- Compare this correlation value against the statistical threshold for the assigned phenotype.
- Label cells falling below the threshold as "unmapped" to indicate potential novel cell types or states.
- Perform downstream analysis on both mapped and unmapped populations to biological similarities and differences between samples.
Protocol for scVI: Probabilistic Integration and Transfer Learning
Model Training and Latent Representation
The scVI workflow begins with data preparation and model training to learn a probabilistic representation of the reference data:
- Format the data into an AnnData object with raw counts and any batch information using scvi-tools.
- Setup the data with
scvi.model.SCVI.setup_anndata(), specifying batch covariates if available. - Initialize the SCVI model:
model = scvi.model.SCVI(adata, use_observed_lib_size=True, dispersion="gene-batch")[114]. - Train the model using
model.train()with default or customized parameters. Training typically requires 100-400 epochs, with early stopping possible to prevent overfitting [112]. - Extract the latent representation using
latent = model.get_latent_representation()and store inadata.obsm["X_scvi"]for downstream analysis.
Transfer Learning and Query Mapping
For transferring annotations from reference to query data:
- Load the pre-trained reference model and prepare the query dataset using the same features as the reference.
- Use
scvi.model.SCVI.load_query_data()to integrate the query data with the reference model, enabling transfer of cell-type annotations and other metadata [114]. - Generate normalized expression values for downstream analysis using
model.get_normalized_expression(). - Perform differential expression analysis between conditions or groups using
model.differential_expression().
The Scientist's Toolkit: Research Reagent Solutions
Essential Materials for scRNA-seq Comparative Analysis
Table 2: Essential Research Reagents and Computational Resources
| Resource | Type | Function in Analysis | Example Sources/Platforms |
|---|---|---|---|
| Reference Datasets | Data | Provide annotated cell populations for training and comparison | Human Protein Atlas [111], Tabula Sapiens [111] [116], CELLxGENE Census [116] |
| Quality Control Tools | Software | Assess cell viability, mitochondrial content, doublets | Scanpy [111], Seurat [117] |
| Batch Correction Algorithms | Algorithm | Mitigate technical variation between datasets | Harmony [117], scVI [112] |
| Normalization Methods | Algorithm | Account for library size differences | Log normalization [117], SCTransform [117] |
| Clustering Methods | Algorithm | Identify cell populations | Leiden algorithm [111], Louvain algorithm [117] |
| Visualization Tools | Software | Project high-dimensional data into 2D/3D space | UMAP [111], t-SNE [117] |
| Differential Expression Tools | Software | Identify statistically significant gene expression changes | scVI [114] [118], traditional statistical tests |
| Cell Type Annotation Databases | Data Resource | Reference for assigning biological identities to clusters | scType [117], manual annotation based on marker genes |
Implementation and Practical Considerations
Successful implementation of these tools requires careful consideration of several practical aspects. For scCompare, the key parameter requiring optimization is the MAD cutoff for determining unmapped cells, which should be calibrated based on the biological context and desired stringency for novel cell type detection [111]. The tool has demonstrated particular utility in detecting distinct cellular populations between different experimental protocols, as evidenced by its application in identifying differences in cardiomyocytes derived from two differentiation protocols [111].
For scVI, important implementation considerations include the use of GPU acceleration for practical runtime with large datasets, careful selection of the latent dimension size (typically 10-30 dimensions), and appropriate handling of the library size parameter [114] [112]. The recent development of scvi-hub further enhances the utility of scVI by providing a repository of pre-trained models that can be directly applied to new query datasets, significantly reducing computational requirements and improving accessibility [116].
Both tools represent valuable additions to the single-cell analysis toolkit, with scCompare offering specialized capabilities for dataset comparison and novelty detection, while scVI provides a unified probabilistic framework for multiple analysis tasks with exceptional scalability. The choice between them should be guided by the specific research questions, dataset characteristics, and analytical priorities of the investigation.
Statistical Methods for Differential Expression and Biomarker Identification
Single-cell RNA sequencing (scRNA-seq) has revolutionized transcriptomic research by enabling the comprehensive analysis of cellular heterogeneity in complex biological systems, providing unprecedented resolution for understanding gene expression dynamics at the individual cell level [46]. Differential expression (DE) analysis serves as a fundamental downstream application of scRNA-seq data, facilitating the identification of biomarker genes for cell type identification and providing critical insights into disease mechanisms [119] [120]. The statistical framework for DE analysis in scRNA-seq must account for unique data characteristics including high-level technical and biological noises, excess overdispersion, low library sizes, sparsity, and a high proportion of zero counts (dropouts) [119] [120]. This application note provides a comprehensive overview of current statistical methodologies, experimental protocols, and analytical frameworks for differential expression and biomarker identification in single-cell transcriptomics, contextualized within a broader thesis on scRNA-seq data analysis research.
Statistical Frameworks for Differential Expression Analysis
Methodological Classification
Current statistical approaches for DE analysis in scRNA-seq data can be distinctly classified into six major categories based on their underlying statistical principles and model architectures [120]. The methodological landscape encompasses generalized linear models (GLM), generalized additive models (GAM), Hurdle models, mixture models, two-class parametric models, and non-parametric approaches, each with specific strengths for addressing particular data challenges [120].
Table 1: Classification of Differential Expression Analysis Methods for scRNA-seq Data
| Method Class | Representative Tools | Underlying Model | Key Features | Limitations |
|---|---|---|---|---|
| GLM-Based | NBID, DECENT, ZINB-WaVE | Negative Binomial, Zero-Inflated NB | Accounts for overdispersion and dropouts | Computational intensity for large datasets |
| Hurdle Models | MAST, MAST-RE | Two-part hierarchical model | Separates dropout vs. expressed states | Assumes independence between two processes |
| Mixture Models | SCDE, DEsingle, scDD | Poisson-Gamma, Zero-Inflated NB | Captures multimodality in expression | Requires spike-ins for technical noise |
| Non-Parametric | Wilcoxon, ROTS, SAMseq | Distribution-free | No assumption of data distribution | Reduced power with small sample sizes |
| Bulk RNA-seq Adapted | DESeq2, edgeR, limma | Negative Binomial, Linear Models | Established methodology | May not handle excess zeros effectively |
| Subject Effect Models | iDESC, muscat-PB, muscat-MM | Mixed models, Pseudo-bulk | Accounts for biological replication | Increased complexity in model specification |
Advanced Considerations for Multi-Subject Designs
In scRNA-seq studies involving multiple subjects, dominant biological variation across individuals presents a significant confounding factor that must be addressed through specialized statistical approaches. Methods such as iDESC (identifying Differential Expression in Single-cell RNA sequencing data with multiple Subjects) utilize a zero-inflated negative binomial mixed model to simultaneously account for subject effects and dropout events [121]. Similarly, muscat provides two distinct approaches: muscat-PB, which aggregates cell-level UMI counts into sample-level "pseudo-bulk" counts analyzed using edgeR, and muscat-MM, which implements generalized linear mixed models (GLMM) on cell-level UMI counts to explicitly model subject variation [121]. These approaches are particularly crucial for distinguishing true disease effects from natural biological variation across subjects in clinical scRNA-seq studies.
Experimental Protocol for Biomarker Identification
Integrated Workflow for scRNA-seq Biomarker Discovery
The following Graphviz diagram illustrates the comprehensive analytical workflow for biomarker identification from single-cell RNA sequencing data, integrating both experimental and computational components:
Sample Preparation and Single-Cell Isolation
The initial critical stage involves generating quality single-cell or single-nuclei suspensions from tissue samples, requiring optimized dissociation protocols that minimize transcriptional stress responses [122]. The decision between single-cell or single-nuclei sequencing depends on research objectives, with single nuclei offering advantages for difficult-to-dissociate tissues or frozen samples, while intact cells provide greater mRNA coverage including cytoplasmic transcripts [122]. Commercially available capture platforms include droplet-based microfluidics (10× Genomics, Illumina), microwell systems (BD Rhapsody, Singleron), and combinatorial barcoding approaches (Scale BioScience, Parse BioScience), each with specific throughput, cost, and input requirements [122].
Analytical Framework Implementation
Following sequencing, raw data undergoes quality control to remove low-quality cells and genes, followed by normalization to account for technical variation [46]. Cell clustering and annotation establishes the cellular taxonomy, enabling cell type-specific differential expression analysis using appropriate statistical methods [120]. Biomarker candidates undergo functional validation through pathway enrichment analysis, regulatory network construction, and experimental confirmation using techniques such as RT-qPCR [123]. A representative application in intellectual disability research identified six ribosomal proteins (RPS27A, RPS21, RPS18, RPS7, RPS5, and RPL9) as hub genes through integrated analysis of single-cell and transcriptomic data [124].
Table 2: Essential Research Reagents and Computational Tools for scRNA-seq Biomarker Studies
| Category | Resource | Specification | Application Context |
|---|---|---|---|
| Cell Capture Platforms | 10× Genomics Chromium | Droplet-based microfluidics | High-throughput cell capture (500-20,000 cells) |
| Illumina (Fluent BioSciences) | Vortex-based droplet capture | Size-agnostic cell capture without microfluidics | |
| BD Rhapsody | Microwell-based capture | Flexible input requirements with large size capacity | |
| Library Preparation | Smart-Seq2 | Full-length transcript protocol | Enhanced sensitivity for low-abundance transcripts |
| Drop-Seq | 3'-end counting with UMIs | High-throughput, low cost per cell | |
| inDrop | 3'-end counting with hydrogel beads | Cost-effective with efficient barcode capture | |
| Analysis Tools | Seurat | R-based toolkit | Comprehensive scRNA-seq analysis pipeline |
| Scanpy | Python-based framework | Scalable analysis for large datasets | |
| iDESC | R package with mixed models | DE analysis with subject effect consideration | |
| DECENT | Zero-inflated model implementation | DE accounting for capture efficiency and dropouts | |
| Reference Databases | Molecular Signatures Database | Curated gene sets | Pathway enrichment and functional analysis |
| STRING | Protein-protein interactions | Network analysis and hub gene identification |
Case Study: Biomarker Identification in Intellectual Disability
A recent investigation demonstrated the integrated application of scRNA-seq and transcriptomic bioinformatics for identifying T cell-specific biomarkers in intellectual disability (ID) [124]. The analysis identified 196 differentially expressed genes through cross-matching of scRNA-seq data and bulk RNA-seq datasets (GSE46831). Functional enrichment analysis revealed significant associations with signal transduction, translation, immune response, and MHC class II-related pathways [124]. Protein-protein interaction network analysis identified six ribosomal proteins (RPS27A, RPS21, RPS18, RPS7, RPS5, and RPL9) as hub genes, with RPS27A emerging as the most significant across eleven topological algorithms [124]. The study additionally identified crucial transcriptional factors (FOXC1, FOXL1, GATA2) and microRNAs (mir-92a-3p, mir-16-5p) as potential regulatory elements, providing a comprehensive molecular framework for understanding ID pathophysiology [124].
The evolving statistical landscape for differential expression analysis in single-cell RNA sequencing data continues to address unique computational challenges including zero-inflation, overdispersion, and biological heterogeneity. Method selection must be guided by experimental design considerations, particularly regarding subject replication, cell numbers, and sequencing depth. Integrated workflows combining appropriate statistical methods with experimental validation provide a robust framework for biomarker discovery across diverse research contexts, from basic biological investigation to clinical translation in disease diagnostics and therapeutic development. As single-cell technologies continue to advance in accessibility and throughput, corresponding developments in analytical methodologies will further enhance resolution and accuracy in biomarker identification.
Strategies for Identifying and Validating Novel Cell States
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological and medical research by enabling the investigation of gene expression at cellular resolution, thereby revealing cellular heterogeneity and diversity [46]. A fundamental task in this domain is the accurate identification of cell states, which represent distinct transcriptional phenotypes within a biological system. Unlike discrete cell types, cell states often exist on a continuous spectrum, representing transitional phases such as differentiation, immune activation, or metabolic adaptation [125]. The ability to precisely identify and validate these states is crucial for understanding developmental biology, tumor microenvironments, and therapeutic responses [126] [46].
The transition from traditional bulk RNA sequencing to scRNA-seq has exposed the complex continuum of cellular phenotypes, where adjacent states exhibit subtle transcriptional differences [125]. This continuum presents significant analytical challenges, as conventional clustering methods often struggle to delineate these finely graded transitions. Furthermore, the high sparsity, technical noise, and batch effects inherent in scRNA-seq data complicate the reliable identification of novel states [127]. This protocol outlines integrated computational and experimental strategies to overcome these challenges, enabling robust discovery and validation of novel cell states within the broader context of single-cell genomics research.
Foundational Concepts and Challenges
Defining Cell States in Single-Cell Data
Cell states represent distinct, often transient, functional or phenotypic conditions within a cell population, characterized by specific gene expression patterns. Unlike stable cell types, states are dynamic and can transition in response to environmental cues, developmental signals, or disease processes. In scRNA-seq data, these states manifest as cellular subpopulations with shared transcriptional profiles that may represent different functional activities, metabolic conditions, or positions along a differentiation trajectory [125].
The identification of these states is complicated by their continuous nature. As scClassify2 highlights, "gene expression states form a continuous space rather than distinct clusters," and "adjacent cell states are typically more similar as they represent transition from one to the other" [125]. This biological reality creates analytical challenges where traditional hard-clustering approaches often fail to capture the nuanced transitions between states.
Technical and Analytical Challenges
Several technical challenges specific to scRNA-seq data affect cell state identification:
- High dimensionality and sparsity: scRNA-seq data typically contain measurements for thousands of genes across thousands of cells, with a high percentage of zero counts due to both biological and technical factors [127].
- Dropout events: Lowly expressed transcripts may not be detected, creating false zeros in the data that obscure true biological signals [127].
- Batch effects: Technical variations between experiments can introduce confounding factors that mimic or mask biological differences [125].
- Transitional state ambiguity: Cells in transitional states often express markers of multiple states simultaneously, creating continuous gradients rather than discrete populations [125].
These challenges necessitate specialized computational approaches that can handle the unique characteristics of single-cell data while capturing the biological reality of continuous cellular phenotypes.
Computational Methodologies
Advanced Clustering Strategies
Traditional hard clustering methods often force cells into discrete categories, oversimplifying the continuous nature of cell states. Recent approaches address this limitation through soft graph clustering techniques that better capture transitional populations.
scSGC (Soft Graph Clustering) represents a significant advancement by addressing limitations of traditional graph-based methods [127]. Unlike "hard graph constructions derived from similarity matrices" that simplify "intercellular relationships into binary edges (0 or 1)," scSGC employs "non-binary edge weights" to characterize "continuous similarities among cells" [127]. This approach more accurately represents the biological continuum of cell states.
The scSGC framework incorporates three innovative components:
- A ZINB-based feature autoencoder to handle sparsity and dropout events in scRNA-seq data
- A dual-channel cut-informed soft graph embedding module that captures continuous similarities between cells
- An optimal transport-based clustering optimization module that achieves optimal delineation of cell populations [127]
Table 1: Comparison of Clustering Approaches for Cell State Identification
| Method | Underlying Principle | Strengths | Limitations |
|---|---|---|---|
| scSGC | Soft graph clustering with non-binary edge weights | Captures continuous cell-state transitions; handles high sparsity | Computational complexity for very large datasets |
| Traditional Hard Clustering | Binary graph constructions with similarity thresholds | Computational efficiency; intuitive parameters | Oversimplifies transitional states; information loss |
| Phenograph | K-nearest neighbor graphs and community detection | Effective for distinct cell populations | Struggles with continuous trajectories |
| Seurat | Graph-based clustering with Louvain/Leiden algorithms | Widely adopted; integrates with comprehensive toolkit | Default parameters may over-split continuous states |
Figure 1: Workflow of soft graph clustering (scSGC) for identifying continuous cell states
Specialized Classification for Sequential States
For identifying adjacent cell states in processes like differentiation or activation, specialized classification approaches that incorporate the inherent ordering of states have shown superior performance.
scClassify2 implements a novel framework specifically designed for "adjacent cell state identification" [125]. Its innovation lies in three key components:
- Transferable biomarker strategies: Using "log-ratio of expression values in multiple samples to capture consistent relationships between two genes" that are more stable across datasets than individual gene expression [125]
- Dual-layer architecture: Integrating "both the expression information and gene co-expression patterns derived from the log-ratio of genes" [125]
- Ordinal regression: Employing a specialized classifier that "captures the sequential nature between transitional cell states" [125]
The dual-layer architecture utilizes a message passing neural network (MPNN) that "incorporates both node and edge information," unlike other graph neural networks that focus primarily on node features [125]. This allows the model to capture subtle gene expression topologies characteristic of closely related cell states.
In benchmark evaluations across eight diverse datasets, scClassify2 achieved a prediction accuracy of 80.76-94.45%, outperforming both its predecessor scClassify (67.22%) and other state-of-the-art methods including scGPT and scFoundation [125].
Spatial Mapping of Cell States
Incorporating spatial context provides critical biological validation for computationally identified cell states, as tissue organization often reflects functional relationships.
CMAP (Cellular Mapping of Attributes with Position) enables "high-resolution mapping of single cells in spatial context" by integrating scRNA-seq data with spatial transcriptomics [128]. This approach addresses the limitation of conventional scRNA-seq, which "inherently sacrifices spatial information, overlooking the pivotal role of extracellular and intracellular interplays in shaping cell fates and function within a tissue context" [128].
The CMAP workflow operates through three progressive mapping stages:
- DomainDivision: Cells are assigned to spatial domains using hidden Markov random field clustering
- OptimalSpot: Cells are aligned to optimal spots/voxels using a cost function and structural similarity index
- PreciseLocation: Exact cellular coordinates are determined using a Spring Steady-State Model [128]
In benchmarking studies, CMAP demonstrated a 73% weighted accuracy in cell location prediction, significantly outperforming CellTrek and CytoSPACE, while achieving a 99% cell usage ratio [128]. This precise spatial mapping enables researchers to validate whether computationally identified cell states occupy biologically plausible tissue locations and to investigate spatial patterns such as immune cell infiltration in tumor microenvironments.
Figure 2: Spatial mapping workflow for validating cell states in tissue context
Experimental Design and Validation
Sample Preparation Considerations
The reliability of cell state identification begins with appropriate experimental design and sample preparation. Critical considerations include:
Cell vs. Nucleus Isolation: The decision to sequence single cells or single nuclei depends on the biological question and tissue characteristics. Single cells capture both nuclear and cytoplasmic mRNA, providing greater transcript detection, while single nuclei are preferable for difficult-to-dissociate tissues or frozen samples [122]. Single nuclei sequencing also enables multiome studies combining transcriptomics with ATAC-seq for chromatin accessibility [122].
Minimizing Dissociation Artifacts: Tissue dissociation can induce stress responses that alter transcriptional profiles. Strategies to mitigate this include:
- Performing digestions on ice to slow transcriptional responses
- Using fixation-based methods like methanol maceration (ACME) or reversible dithio-bis(succinimidyl propionate) fixation
- Implementing fluorescence-activated cell sorting with live/dead stains to eliminate debris [122]
Platform Selection: Different scRNA-seq platforms offer distinct advantages. Full-length transcript methods (Smart-Seq2, MATQ-Seq) excel at detecting isoforms and low-abundance genes, while 3'-end counting methods (10x Genomics, Drop-Seq) enable higher throughput and lower cost per cell [46]. The choice should align with the specific goals of cell state identification.
Table 2: Single-Cell Platform Selection for Cell State Identification
| Platform | Transcript Coverage | UMIs | Cell Throughput | Best Suited for Cell State Analysis |
|---|---|---|---|---|
| Smart-Seq2 | Full-length | No | Low | Detecting low-abundance transcripts; isoform usage |
| 10x Genomics | 3'-end | Yes | High | Large-scale state characterization; rare cell populations |
| MATQ-Seq | Full-length | Yes | Medium | Increased accuracy in transcript quantification |
| Seq-Well | 3'-only | Yes | High | Portable applications; challenging sample types |
Analytical Validation Framework
Robust validation of novel cell states requires multiple lines of evidence:
Genomic Validation: Copy number variation (CNV) analysis can distinguish malignant from non-malignant cells and identify subclonal populations. As demonstrated in ER+ breast cancer studies, "higher CNV scores in tumor cells from metastatic patient samples" correlate with disease progression and can help define aggressive cell states [126]. Tools like InferCNV and CaSpER enable CNV inference from scRNA-seq data [126].
Marker Gene Validation: Identified cell states should exhibit consistent marker gene expression. For example, in breast cancer metastasis, "CCL2+ macrophages, exhausted cytotoxic T cells, and FOXP3+ regulatory T cells" defined pro-tumor microenvironment states, while "FOLR2 and CXCR3 positive macrophages" were associated with primary tumors [126]. These molecular signatures provide validating evidence for state definitions.
Functional Validation: Pathway enrichment analysis of state-specific genes can reveal functional correlates. In primary breast cancer, "increased activation of the TNF-α signaling pathway via NF-κB" represented a potential therapeutic target state [126]. Such functional annotations strengthen the biological relevance of identified states.
Essential Research Tools
Computational Toolkit
Table 3: Essential Computational Tools for Cell State Identification
| Tool | Primary Function | Key Features | Application in Cell State Analysis |
|---|---|---|---|
| scClassify2 | Cell state classification | Ordinal regression for sequential states; dual-layer MPNN architecture | Identification of adjacent cell states in differentiation or activation |
| scSGC | Soft graph clustering | Non-binary edge weights; ZINB-based autoencoder | Capturing continuous transitions between cell states |
| CMAP | Spatial mapping | Three-level mapping; precise coordinate assignment | Validating spatial organization of cell states |
| scDSC | Deep embedded clustering | Graph neural network with mutual supervision | Integrating feature learning and clustering optimization |
| scGPT | Foundation model | Pre-trained on massive single-cell datasets | Transfer learning for cell state annotation |
| scran | Normalization | Pool-based size factor calculation | Data preprocessing for accurate state identification |
Visualization and Interpretation
Effective visualization is crucial for interpreting and communicating cell state analyses, particularly given the complexity and multidimensionality of the data.
scatterHatch addresses visualization challenges by creating "colorblind-friendly scatter plots by redundant coding of cell groups using colors as well as patterns" [129]. This approach is particularly valuable for distinguishing closely related cell states in dimensional reduction plots (UMAP, t-SNE), where traditional color-only coding may be insufficient to convey subtle distinctions, especially for the 8% of males and 0.5% of females with color vision deficiencies [129].
The package combines "40 high-contrast CVD-friendly colors" with six default patterns (horizontal, vertical, diagonal, checkers, etc.) to create visually distinct representations for up to 280 different groups [129]. This enhanced visualization capability supports more accurate interpretation of cell state relationships and facilitates inclusive science communication.
Integrated Protocol for Cell State Identification
This section provides a step-by-step protocol for implementing the described strategies in a coordinated workflow for novel cell state identification and validation.
Sample Preparation and Sequencing
- Tissue Dissociation: Prepare single-cell suspensions using optimized dissociation protocols appropriate for your tissue type. Consider fixation methods if processing time or sample stress is a concern [122].
- Quality Control: Assess cell viability and integrity using microscopy and flow cytometry. Aim for >80% viability in cell suspensions [122].
- Platform Selection: Choose an appropriate scRNA-seq platform based on your target cell numbers, sequencing depth requirements, and budget [46].
- Library Preparation: Follow manufacturer protocols for your selected platform. Include appropriate UMIs to correct for amplification biases [46].
- Sequencing Depth: Target approximately 20,000-50,000 reads per cell to balance cost and transcript detection sensitivity [122].
Computational Analysis Pipeline
Data Preprocessing:
- Perform quality control filtering to remove low-quality cells and doublets
- Normalize using methods appropriate for your data (e.g., scran, SCTransform)
- Integrate datasets if multiple samples are included [127]
Cell State Identification:
Satial Validation:
- Apply CMAP to map identified states to spatial coordinates if spatial transcriptomics data is available
- Validate whether computationally identified states correspond to spatially distinct regions [128]
Functional Annotation:
- Perform pathway enrichment analysis on state-specific genes
- Infer cell-cell communication patterns using tools like CellChat or NicheNet [126]
Experimental Validation
Orthogonal Validation: Confirm key cell states using alternative methods such as:
- Multiplexed fluorescence in situ hybridization (FISH)
- Flow cytometry with state-specific marker panels
- Immunofluorescence staining [126]
Functional Assays: Design experiments to test functional properties of identified states, such as:
- Drug response assays for therapeutic vulnerability states
- Migration or invasion assays for metastatic states
- Cytokine production assays for immune activation states [126]
The identification and validation of novel cell states requires an integrated approach combining sophisticated computational methods with careful experimental design and orthogonal validation. The strategies outlined in this protocol leverage recent advances in soft clustering, ordinal classification, and spatial mapping to address the unique challenges posed by the continuous nature of cellular phenotypes. As single-cell technologies continue to evolve, these methodologies will enable researchers to extract increasingly nuanced understanding of cellular heterogeneity in development, homeostasis, and disease.
Conclusion
Single-cell RNA sequencing has irrevocably transformed our ability to probe cellular heterogeneity, driving advances in basic biology and precision medicine. The integration of robust foundational workflows with advanced machine learning methodologies is key to unlocking the full potential of scRNA-seq data. While challenges related to data noise, integration, and interpretation persist, ongoing developments in computational tools and best practices are steadily overcoming these hurdles. Future progress hinges on enhanced model interpretability, improved multi-omics integration, and the establishment of standardized frameworks for clinical translation. As the field continues to mature, scRNA-seq analysis will undoubtedly play an increasingly central role in identifying novel therapeutic targets, understanding disease mechanisms, and ultimately guiding personalized treatment strategies, solidifying its place as an indispensable tool in modern biomedical research.
References
- 1. - Single methods explained | INTEGRA cell RNA sequencing [https://www.integra-biosciences.com/global/en/blog/article/single-cell-rna-sequencing-methods-plate-droplet-and-microwell-based-approaches]
- 2. Getting Started with scRNA-seq: Experimental Design and ... [https://www.parsebiosciences.com/blog/getting-started-with-scrna-seq-experimental-design-and-sample-preparation/]
- 3. Single-Cell RNA-Seq Workshop 2025 [https://www.ecseq.com/workshops/workshop_2025-08-Single-Cell-RNA-Seq-Data-Analysis]
- 4. - Droplet - based : decoding cellular... single cell RNA sequencing [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06996-0]
- 5. Experimental design for single-cell RNA sequencing [https://pubmed.ncbi.nlm.nih.gov/29126257/]
- 6. A simplified preparation method for single -nucleus RNA - sequencing ... [https://www.nature.com/articles/s41598-025-97053-9?error=cookies_not_supported&code=c5dd8ffa-26c1-4ad1-b7cd-2ef43fb6978e]
- 7. Getting Started with scRNA-seq: Post-Sequencing Data ... [https://www.parsebiosciences.com/blog/getting-started-with-scrna-seq-post-sequencing-data-analysis/]
- 8. Current best practices in single‐cell RNA‐seq analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 9. GEO Accession viewer [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM8842803]
- 10. Powerful scRNA-seq Data Analysis Tools in 2025 [https://www.nygen.io/resources/blog/single-cell-scrna-seq-multi-omics-data-analysis-tools]
- 11. Model-based dimensionality reduction for single-cell RNA- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12342792/]
- 12. Model-based understanding of single-cell CRISPR screening [https://www.nature.com/articles/s41467-019-10216-x]
- 13. Comprehensive generation, visualization, and reporting of ... [https://www.nature.com/articles/s41467-022-29212-9]
- 14. Common Considerations for Quality Control Filters ... [https://www.10xgenomics.com/analysis-guides/common-considerations-for-quality-control-filters-for-single-cell-rna-seq-data]
- 15. Comparison of single-cell RNA-seq methods to enable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539239/]
- 16. Best Practices for Analysis of 10x Genomics Single Cell ... [https://www.10xgenomics.com/analysis-guides/best-practices-analysis-10x-single-cell-rnaseq-data]
- 17. Evaluating cell-specific gene expression using single ... [https://www.nature.com/articles/s41598-025-21595-1]
- 18. Data normalization for addressing the challenges in the ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10364-5]
- 19. 9. Dimensionality Reduction [https://www.sc-best-practices.org/preprocessing_visualization/dimensionality_reduction.html]
- 20. A Comparison for Dimensionality Reduction Methods of ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.646936/full]
- 21. How single cell sequencing data analysis works [https://www.10xgenomics.com/blog/how-single-cell-sequencing-data-analysis-works]
- 22. Single-cell RNA-seq Data Normalization [https://www.10xgenomics.com/analysis-guides/single-cell-rna-seq-data-normalization]
- 23. Feature selection methods affect the performance of ... [https://www.nature.com/articles/s41592-025-02624-3]
- 24. 8. Feature selection [https://www.sc-best-practices.org/preprocessing_visualization/feature_selection.html]
- 25. A practical guide to single-cell RNA-sequencing for ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-017-0467-4]
- 26. GUIDING CLUSTERING AND ANNOTATION IN SINGLE ... [https://pubmed.ncbi.nlm.nih.gov/40654835/]
- 27. Navigating single-cell RNA-sequencing: protocols, tools ... [https://genomicsinform.biomedcentral.com/articles/10.1186/s44342-025-00044-5]
- 28. Establishing single cell RNA transcriptomics: a brief guide [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403637/]
- 29. Flexible experimental designs for valid single-cell RNA ... [https://www.nature.com/articles/s41467-020-16905-2]
- 30. 6 Basic Quality Control (QC) and Exploration of scRNA-seq ... [https://www.singlecellcourse.org/basic-quality-control-qc-and-exploration-of-scrna-seq-datasets.html]
- 31. Global trends in machine learning applications for single-cell ... [https://hereditasjournal.biomedcentral.com/articles/10.1186/s41065-025-00528-y]
- 32. Bioinformatics perspectives on transcriptomics [https://www.rna-seqblog.com/bioinformatics-perspectives-on-transcriptomics-a-comprehensive-review-of-bulk-and-single-cell-rna-sequencing-analyses/]
- 33. Single‑cell RNA sequencing data dimensionality reduction ... [https://www.spandidos-publications.com/10.3892/wasj.2025.315]
- 34. Trajectory inference from single-cell genomics data with a ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012752]
- 35. Exploring machine learning strategies for single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12291542/]
- 36. Benchmarking of dimensionality reduction methods to ... [https://www.nature.com/articles/s41598-025-12021-7]
- 37. Infusing structural assumptions into dimensionality ... [https://www.nature.com/articles/s42003-025-07872-9]
- 38. Compositional data modeling of high-dimensional single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12539173/]
- 39. Clinical application of single‐cell RNA sequencing in disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576031/]
- 40. Single Cell Sequencing and The Future of Drug Discovery [https://www.parsebiosciences.com/blog/single-cell-sequencing-and-the-future-of-drug-discovery/]
- 41. Single-cell RNA sequencing identifies potential critical ... [https://www.nature.com/articles/s41598-025-23975-z]
- 42. Integrating single-cell RNA sequencing and artificial ... [https://www.nature.com/articles/s41698-025-00952-3]
- 43. Applications and emerging challenges of single-cell RNA ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000030]
- 44. Single-cell transcriptomics reveals biomarker ... [https://www.nature.com/articles/s41523-025-00803-1]
- 45. Single-Cell Sequencing: An Emerging Tool for Biomarker ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12153836/]
- 46. Navigating single-cell RNA-sequencing: protocols, tools, databases, and applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12085826/]
- 47. Exploration and Application of Malignant Cell ... [https://pubmed.ncbi.nlm.nih.gov/40768639/]
- 48. Single-Cell Informatics for Tumor Microenvironment and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11050520/]
- 49. Data analysis guidelines for single-cell RNA-seq in ... [https://mmrjournal.biomedcentral.com/articles/10.1186/s40779-022-00434-8]
- 50. Characterizing the tumor microenvironment at the single- ... [https://www.nature.com/articles/s41413-022-00237-6]
- 51. Harnessing Single-Cell RNA-Seq for Computational Drug Repurposing in Cancer Immunotherapy [https://www.mdpi.com/1424-8247/18/11/1769]
- 52. Single-cell network biology for resolving cellular ... [https://www.nature.com/articles/s12276-020-00528-0]
- 53. Single-cell RNA sequencing highlights the unique tumor microenvironment of small cell neuroendocrine cervical carcinoma - PubMed [https://pubmed.ncbi.nlm.nih.gov/39762893/]
- 54. Single-cell sequencing to multi-omics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11438019/]
- 55. Integrating scRNA-seq and scATAC-seq data [https://satijalab.org/seurat/articles/seurat5_atacseq_integration_vignette]
- 56. A guide to multi-omics data collection and integration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9747357/]
- 57. Multiomics data integration to reveal chromatin remodeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9579702/]
- 58. INSTINCT: Multi-sample integration of spatial chromatin ... [https://www.nature.com/articles/s41467-025-56535-0]
- 59. Single-cell multiomics data integration and generation with ... [https://www.sciencedirect.com/science/article/pii/S2667237525002474]
- 60. Tutorial: Multi-omics data visualization with PathVisio [https://pathvisio.org/tutorials/multi-omics-tutorial.html]
- 61. Visual analysis of multi-omics data [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2024.1395981/full]
- 62. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 63. Applications of single-cell RNA sequencing in drug ... [https://www.nature.com/articles/s41573-023-00688-4]
- 64. Dissecting crosstalk induced by cell-cell communication ... [https://www.nature.com/articles/s41467-025-61149-7]
- 65. Single-Cell RNA Sequencing in Drug Discovery and ... [https://rna.cd-genomics.com/resource/scrna-seq-drug-discovery.html]
- 66. Exploring and mitigating shortcomings in single-cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03525-6]
- 67. Single cell analytic tools for drug discovery and development [https://pmc.ncbi.nlm.nih.gov/articles/PMC4883669/]
- 68. RNA Sequencing in Drug Discovery and Development [https://www.lexogen.com/blog/rna-sequencing-in-drug-discovery-and-development/]
- 69. Imputing single-cell RNA-seq data by considering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8035992/]
- 70. Introduction to Ambient RNA Correction [https://www.10xgenomics.com/analysis-guides/introduction-to-ambient-rna-correction]
- 71. Decontamination of ambient RNA in single-cell RNA-seq with ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-1950-6]
- 72. Regulatory network-based imputation of dropouts in single ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009849]
- 73. Evaluating imputation methods for single-cell RNA-seq data [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05417-7]
- 74. An efficient scRNA-seq dropout imputation method using ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04493-x]
- 75. Embracing the dropouts in single-cell RNA-seq analysis [https://www.nature.com/articles/s41467-020-14976-9]
- 76. Mitigating ambient RNA and doublets effects on single cell ... [https://www.sciencedirect.com/science/article/abs/pii/S0304383525002599]
- 77. A contamination focused approach for optimizing the single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10366499/]
- 78. Evaluating the performance of dropout imputation and ... [https://www.sciencedirect.com/science/article/pii/S0010482522004802]
- 79. Filtering cells with high mitochondrial content depletes viable ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03559-w]
- 80. Single-Cell RNA sequencing reveals mitochondrial ... [https://www.nature.com/articles/s41598-025-85169-x]
- 81. Guidelines for mitochondrial RNA analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11292373/]
- 82. Guidelines for mitochondrial RNA analysis [https://www.sciencedirect.com/science/article/pii/S2162253124001495]
- 83. Single-cell RNA-sequencing reveals early mitochondrial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12089458/]
- 84. Batch correction methods used in single-cell RNA sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315870/]
- 85. Integrating single-cell RNA-seq datasets with substantial ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12126-3]
- 86. Reference-informed evaluation of batch correction for ... [https://www.nature.com/articles/s42003-025-07947-7]
- 87. Guide to Getting Started with 10X Genomics Single Cell RNA ... [https://biotech.wisc.edu/guide-to-getting-started-with-10x-genomics-single-cell-rna-seq/]
- 88. Design and computational analysis of single-cell RNA ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0927-y]
- 89. Optimization of clustering parameters for single-cell RNA ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1562410/full]
- 90. BestopCloud: an integrated one-stop solution for single-cell ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12072-0]
- 91. scICE: enhancing clustering reliability and efficiency of ... [https://www.nature.com/articles/s41467-025-60702-8]
- 92. Tuning parameters of dimensionality reduction methods for ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02128-7]
- 93. scVGAMF: a novel imputation method for scRNA-seq data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12554638/]
- 94. PCLDA: An interpretable cell annotation tool for single- ... [https://www.sciencedirect.com/science/article/pii/S2001037025002818]
- 95. Cell Preparation for Single Cell Protocols [https://www.10xgenomics.com/support/universal-five-prime-gene-expression/documentation/steps/sample-prep/single-cell-protocols-cell-preparation-guide]
- 96. Mapping and Validation of scRNA-Seq-Derived Cell-Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9096838/]
- 97. Prioritization and functional validation of target genes from ... [https://www.nature.com/articles/s42003-023-05006-7]
- 98. How To Validate Single-Cell RNA-Seq Data? - CD Genomics [https://www.spatial-omicslab.com/resource/validate-single-cell-rna-seq.html]
- 99. Protocol for interpretable and context-specific single-cell- ... [https://www.sciencedirect.com/science/article/pii/S2666166725000760]
- 100. Tahoe Open Sources Tahoe-100M, the World's Largest ... [https://arcinstitute.org/news/arc-vevo]
- 101. Public RNA-Seq and scRNA-Seq Databases [https://bigomics.ch/blog/ultimate-guide-to-public-rnaseq-and-sc-rna-seq-databases/]
- 102. Arc Virtual Cell Atlas launches, combining data from over ... [https://www.rna-seqblog.com/arc-virtual-cell-atlas-launches-combining-data-from-over-300-million-cells/]
- 103. Bulk RNA-seq and data analysis [https://www.protocols.io/view/bulk-rna-seq-and-data-analysis-dsev6be6.html]
- 104. Which single-cell analysis tool is best? Scientists offer advice [https://www.nature.com/articles/d41586-022-04426-5]
- 105. Comparative benchmarking of single-cell clustering ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03719-y]
- 106. Benchmarking scRNA-seq copy number variation callers [https://www.nature.com/articles/s41467-025-62359-9]
- 107. Performance comparison of high throughput single-cell ... [https://www.sciencedirect.com/science/article/pii/S2405844024132161]
- 108. Benchmarking full-length transcript single cell mRNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-09014-5]
- 109. Benchmarking single-cell RNA-sequencing protocols for ... [https://www.nature.com/articles/s41587-020-0469-4]
- 110. Benchmarking Single-Cell RNA Sequencing Protocols for Cell ... [https://explore.data.humancellatlas.org/projects/6e177195-0ac0-468b-99a2-87de96dc9db4]
- 111. A Strategy to Compare Single-Cell RNA Sequencing Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11457179/]
- 112. Deep Generative Modeling for Single-cell Transcriptomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC6289068/]
- 113. A Strategy to Compare Single-Cell RNA Sequencing Data ... [https://www.bluerocktx.com/a-strategy-to-compare-single-cell-rna-sequencing-data-sets-provides-phenotypic-insight-into-cellular-heterogeneity-underlying-biological-similarities-and-differences-between-samples/]
- 114. single-cell Variational Inference; Python class [https://docs.scvi-tools.org/en/1.3.3/user_guide/models/scvi.html]
- 115. A Strategy to Compare Single-Cell RNA Sequencing Data ... [https://pubmed.ncbi.nlm.nih.gov/39376245/]
- 116. Scvi-hub: an actionable repository for model-driven single ... [https://www.nature.com/articles/s41592-025-02799-9]
- 117. Best single-cell RNA-sequencing data analysis tools in 2024 [https://www.biomage.net/blog/best-single-cell-rna-sequencing-data-analysis-tools-in-2024]
- 118. scRNA-seq [https://docs.scvi-tools.org/en/1.3.3/tutorials/index_scrna.html]
- 119. A Comprehensive Survey of Statistical Approaches for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8701051/]
- 120. Differential Expression Analysis of Single-Cell RNA-Seq Data [https://www.mdpi.com/1099-4300/24/7/995]
- 121. iDESC: identifying differential expression in single-cell RNA ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05432-8]
- 122. Establishing single cell RNA transcriptomics: a brief guide [https://frontiersinzoology.biomedcentral.com/articles/10.1186/s12983-025-00579-x]
- 123. Portfolio analysis of single-cell RNA-sequencing and ... [https://pubmed.ncbi.nlm.nih.gov/41246299/]
- 124. Identification of cell specific biomarkers for intellectual ... [https://www.nature.com/articles/s41598-025-85162-4]
- 125. A message passing framework for precise cell state ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03722-3]
- 126. Single-cell RNA sequencing reveals different cellular ... [https://www.nature.com/articles/s41523-025-00808-w]
- 127. Soft graph clustering for single-cell RNA sequencing data [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06231-z]
- 128. High-resolution mapping of single cells in spatial context [https://www.nature.com/articles/s41467-025-61667-4]
- 129. Generating colorblind-friendly scatter plots for single-cell data [https://elifesciences.org/articles/82128]