Benchmarking Genome Assemblers: A Comprehensive Guide for Biomedical Researchers
This article provides a comprehensive guide to benchmarking genome assemblers, a critical step in genomics that directly impacts downstream applications in drug development and clinical research.
Benchmarking Genome Assemblers: A Comprehensive Guide for Biomedical Researchers
Abstract
This article provides a comprehensive guide to benchmarking genome assemblers, a critical step in genomics that directly impacts downstream applications in drug development and clinical research. We explore the foundational principles of assembly evaluation, detail methodological approaches for long-read and hybrid sequencing data, and present strategies for troubleshooting and optimization. By synthesizing findings from recent large-scale benchmarks, we offer a validated framework for selecting assembly tools and pipelines, empowering scientists to generate high-quality genomic resources essential for uncovering disease mechanisms and advancing personalized medicine.
The Genome Assembly Landscape: Why Benchmarking is Crucial for Accurate Genomics
The reliability of genome assemblies is a foundational element in modern genomic research, acting as the primary scaffold upon which all subsequent biological interpretations are built. The quality of a genome assembly directly controls the fidelity of functional annotation and the accuracy of comparative genomics analyses, which in turn influences downstream applications in drug development and disease mechanism studies. Research has demonstrated that assemblies with different qualities can lead to markedly different biological conclusions, making rigorous quality assessment a non-negotiable step in genomic workflows [1] [2].
The principle of "Garbage In, Garbage Out" is particularly pertinent to genome assembly. Errors in the assembly—whether at the base level, such as single-nucleotide inaccuracies, or the structural level, including misjointed contigs or missing regions—cascade through all downstream analyses. These errors can manifest as missing exons, fragmented genes, incorrectly inferred evolutionary relationships, or entirely missed genetic variants of clinical importance [3]. For researchers and drug development professionals, this translates to potential misinterpretations of a gene's functional role, an organism's pathogenic mechanism, or the identification of flawed drug targets. Therefore, a comprehensive understanding of how to assess assembly quality and its subsequent impact is crucial for ensuring the integrity of genomic research.
Assessing Genome Assembly Quality: The 3C Principles and Benchmarking Tools
The quality of a genome assembly is quantitatively assessed based on three core principles, often called the "3Cs": Contiguity, Completeness, and Correctness [3].
- Contiguity measures how much of the genome is reconstructed into long, uninterrupted stretches. Key metrics include the N50/L50, where a higher N50 value indicates a more contiguous assembly, and the total number of contigs or scaffolds, where a lower number is preferable.
- Completeness evaluates what proportion of the actual genome is present in the assembly. This is assessed by looking for a core set of universal single-copy orthologs using tools like BUSCO, where a score above 95% is considered good, or by analyzing k-mer spectra from raw reads to see what fraction is represented in the assembly [4] [3].
- Correctness gauges the accuracy of each base pair and the overall structure. Base-level correctness can be checked by mapping high-quality short reads to the assembly, while structural correctness often requires comparison to a known reference or the use of technologies like Hi-C or Bionano [3].
To streamline this multi-faceted evaluation, several integrated tools have been developed. QUAST provides a comprehensive report on assembly metrics with or without a reference genome. GenomeQC is an interactive web framework that integrates a suite of quantitative measures, including BUSCO for gene space completeness and the LTR Assembly Index (LAI) for assessing the completeness of repetitive regions, which is particularly valuable for plant genomes [4]. The Genome Assembly Evaluation Pipeline (GAEP) is another comprehensive tool that utilizes NGS data, long-read data, and transcriptome data to evaluate assemblies for continuity, accuracy, completeness, and redundancy [3].
Table 1: Key Tools for Genome Assembly Quality Assessment
| Tool | Primary Function | Key Metrics | Notable Features |
|---|---|---|---|
| QUAST | Quality Assessment Tool for Genome Assemblies | N50, misassemblies, mismatches per 100 kbp | Works with/without reference genome; user-friendly reports [3]. |
| GenomeQC | Integrated Quality Assessment | NG(X) plots, BUSCO, LAI, contamination check | Web framework; assesses both assembly and gene annotation [4]. |
| BUSCO | Benchmarking Universal Single-Copy Orthologs | Complete, fragmented, missing orthologs (%) | Measures gene space completeness against conserved gene sets [4] [3]. |
| GAEP | Genome Assembly Evaluation Pipeline | Basic stats, BUSCO, k-mer analysis | Uses multiple data sources (NGS, long-read, transcriptome) for evaluation [3]. |
| Merqury | K-mer-based Evaluation | QV, k-mer completeness | Uses k-mer spectra to assess base-level accuracy and completeness [5]. |
Experimental Protocols for Benchmarking Assemblers
To objectively compare the performance of different genome assemblers, a standardized benchmarking approach is essential. The following protocol, synthesized from recent large-scale studies, outlines a robust methodology.
Experimental Design and Data Preparation
The foundation of a reliable benchmark is the use of well-characterized reference samples and a variety of sequencing data. The Genome in a Bottle (GIAB) Consortium provides widely adopted reference materials, such as the human sample HG002 [5]. For a comprehensive benchmark, data from multiple sequencing technologies should be incorporated:
- Long-read data from Oxford Nanopore Technologies (ONT) or PacBio to resolve repetitive regions and improve contiguity.
- Short-read data from Illumina for high base-level accuracy and polishing.
- The data should be subsampled to various coverages (e.g., 30X, 50X) and read lengths to evaluate performance under different data constraints [1].
Assembly and Polishing Workflow
A typical benchmarking workflow involves multiple stages:
- Assembly: The selected assemblers are run on the designated sequencing datasets. As highlighted in a 2025 benchmark, both long-read-only assemblers (e.g., Flye, Canu) and hybrid assemblers (e.g., MaSuRCA) should be tested [5] [6].
- Polishing: The initial draft assemblies are refined through iterative polishing. The benchmark found that the best results were often achieved with a combination of long-read-based polishing (e.g., Racon) followed by short-read-based polishing (e.g., Pilon) [5].
- Scaffolding (optional): For chromosome-level assemblies, Hi-C data can be used with scaffolding tools like SALSA2 or ALLHIC [1].
Quality Assessment and Analysis
The final, polished assemblies are evaluated using the metrics and tools described in Section 2. A comprehensive analysis includes:
- QUAST for contiguity and misassembly statistics.
- BUSCO for gene completeness.
- Merqury for k-mer-based quality valuation (QV) and completeness [5].
- LAI for assessing repetitive region assembly in complex genomes [4]. The results are then compiled for cross-assembler comparison.
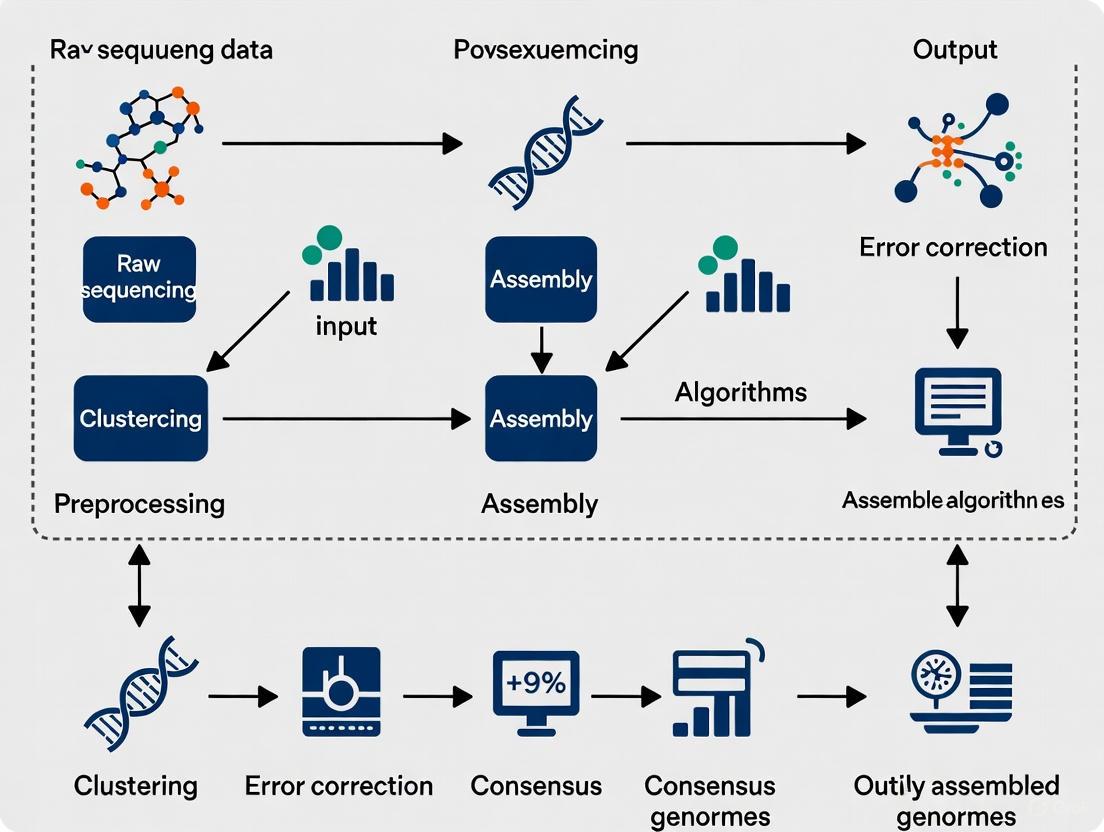
Diagram 1: Genome Assembler Benchmarking Workflow. This flowchart outlines the key experimental stages for objectively comparing genome assemblers, from data preparation to final analysis.
Quantitative Comparison of Genome Assemblers
Recent benchmarking studies provide critical quantitative data on the performance of modern assemblers. A 2025 study evaluating 11 pipelines for hybrid de novo assembly of human genomes using ONT and Illumina data found that Flye outperformed other assemblers, especially when ONT reads were pre-corrected with tools like Ratatosk [5]. The study further demonstrated that polishing is a non-negotiable step, with the best results coming from two rounds of Racon followed by Pilon, which significantly improved both assembly accuracy and continuity [5].
Table 2: Benchmarking Results of Assembly and Polishing Pipelines (Adapted from [5])
| Assembly Strategy | Best-Performing Tool | Key Quality Metrics (Post-Polishing) | Computational Cost |
|---|---|---|---|
| Long-Read (ONT) Assembly | Flye | High continuity (N50), superior BUSCO completeness | Moderate |
| Hybrid Assembly | MaSuRCA | Good balance of continuity and base accuracy | High |
| Pre-Assembly Correction | Ratatosk + Flye | Improved assembly continuity and accuracy | Very High |
| Polishing Strategy | Racon (2x) + Pilon (1x) | Optimal baseline and structural variant accuracy | Moderate |
The impact of input data quality and assembly strategy was further explored in a 2021 study on a non-model plant genome. It revealed that data subsampled for longer read lengths, even at lower coverage, produced more contiguous and complete assemblies than data with shorter reads but higher coverage [1]. This finding underscores the critical importance of read length for resolving complex genomic regions. The study also highlighted that the success of downstream scaffolding with Hi-C data is heavily dependent on the underlying contig assembly being accurate; problems in the initial assembly cannot be resolved by Hi-C and may even be exacerbated [1].
Impact of Assembly Quality on Functional Annotation
Functional annotation is the process of attaching biological information—such as gene predictions, functional domains, and Gene Ontology (GO) terms—to a genome sequence. The quality of the underlying assembly is the primary determinant of annotation accuracy and completeness. A fragmented or erroneous assembly directly leads to fragmented or missing gene models, mis-identified exon-intron boundaries, and ultimately, an incomplete or misleading functional catalog of the organism [6] [3].
A case study on the pathogenic protozoan Balamuthia mandrillaris vividly illustrates this dependency. Researchers performed a hybrid assembly using both Illumina short reads and ONT long reads, resulting in a genome with superior assembly metrics compared to previously available drafts. This high-quality assembly enabled a comprehensive functional annotation, which successfully identified 11 out of 15 genes that had previously been described as potential therapeutic targets. This was only possible because the improved assembly provided a more complete and accurate genomic context [6]. In contrast, an assembly littered with gaps and misassemblies will cause gene prediction algorithms to fail, leaving researchers with an incomplete picture of the organism's biology and potentially missing critical virulence factors or drug targets.
Impact of Assembly Quality on Comparative Genomics
Comparative genomics relies on the accurate comparison of genomic features across different species or strains to infer evolutionary relationships, identify conserved regions, and discover genes underlying specific traits. The foundation of these analyses is a set of high-quality, colinear genome sequences. Errors in individual assemblies propagate through comparative analyses, leading to incorrect inferences of gene gain and loss, flawed phylogenetic trees, and misidentification of genomic rearrangements [7].
For example, a core analysis in comparative genomics is the definition of the pangenome, which comprises the core genome (genes shared by all strains) and the accessory genome (genes present in some strains). If one assembly in a multi-species comparison is highly fragmented, genes may be split across multiple contigs or missed entirely. This would artificially inflate the number of "unique" genes in the accessory genome for that species while simultaneously shrinking the core genome, leading to a distorted view of evolutionary relationships and functional conservation [8] [7]. The PATRIC database, as a bacterial bioinformatics resource center, exemplifies the need for "virtual integration" of high-quality, uniformly annotated genomes to enable reliable comparative studies [8]. Consistent, high-quality assemblies are therefore prerequisite for meaningful comparative genomics that can accurately trace the evolution of pathogenicity or antibiotic resistance across bacterial lineages.
Table 3: Key Research Reagent Solutions for Genome Assembly and Annotation
| Resource / Tool | Type | Function in Research |
|---|---|---|
| GIAB Reference Materials | Biological Standard | Provides benchmark genomes (e.g., HG002) for validating assembly and variant calling accuracy [5]. |
| PATRIC | Bioinformatics Database | An all-bacterial bioinformatics resource center for comparative genomic analysis with integrated tools [8]. |
| Flye | Software | A long-read assembler that has demonstrated top performance in benchmarks for continuity and completeness [5]. |
| Racon & Pilon | Software | A combination of polishers used to correct base-level errors in a draft assembly using long and short reads, respectively [5]. |
| BUSCO Dataset | Software/Database | A set of universal single-copy orthologs used to quantitatively assess the completeness of a genome assembly [4] [3]. |
| Funannotate | Software | A pipeline for functional annotation of a genome, integrating gene prediction, functional assignment, and non-coding RNA identification [6]. |
| Restauro-G | Software | A rapid, automated genome re-annotation system for bacterial genomes, ensuring consistent annotation across datasets [9]. |
The body of evidence from systematic benchmarks and case studies leads to an unequivocal conclusion: the quality of a genome assembly is not a mere technical detail but a fundamental variable that dictates the success of all downstream genomic analyses. Investments in superior sequencing data (particularly long reads), robust assembly algorithms like Flye, and rigorous polishing protocols yield dividends in the form of more complete and accurate functional annotations and more reliable comparative genomic insights. For researchers and drug developers, prioritizing genome quality is a critical step toward ensuring that biological discoveries and therapeutic target identification are built upon a solid and trustworthy foundation.
The quality of a genome assembly is fundamental, as it directly impacts all subsequent biological interpretations and analyses [10]. The assessment of this quality is universally structured around three core dimensions: contiguity, completeness, and correctness—collectively known as the "3 Cs" [10] [3] [11]. Relying on a single metric, particularly those related only to contiguity like the popular N50, is a common but misleading practice. High contiguity does not guarantee an accurate assembly; in fact, the most contiguous assembly may also be the most incorrect if misjoins have artificially inflated contig sizes [12] [11]. A holistic evaluation is therefore indispensable. This guide provides a structured overview of the core metrics and methodologies for evaluating genome assemblies, framing them within the context of benchmarking genome assemblers. It is designed to help researchers and developers objectively compare assembler performance by synthesizing current evaluation protocols and experimental data.
Defining the Core Metrics: The "3 Cs" Framework
An ideal genome assembly is highly contiguous, complete, and correct. These three principles serve as the pillars for a robust assessment, though they can often be contradictory, as optimizing for one can sometimes come at the expense of another [3]. The following sections define and detail the metrics associated with each "C."
Contiguity
Contiguity measures how well an assembly reconstructs long, uninterrupted DNA sequences, reflecting the effectiveness of the assembly process in extending sequences without breaks [3] [11]. It is primarily concerned with the size and number of the assembled fragments.
- N50 / L50: The N50 is defined as the length of the shortest contig or scaffold such that contigs of this length or longer contain 50% of the total assembly length [10] [3]. The L50 is the corresponding number of contigs. In the era of long-read sequencing, a contig N50 over 1 Mb is often considered good for many eukaryotic genomes [10]. A major critique of N50 is that it can be artificially inflated by misassemblies, making it an unreliable standalone metric [11].
- NG50 / LG50: These metrics are analogous to N50 and L50 but are calculated with respect to the estimated genome size rather than the assembly size. This prevents a larger but incomplete assembly from appearing more contiguous than a smaller, more complete one.
- Number of contigs/scaffolds and gaps: The total count of contigs and scaffolds, along with the number and length of gaps between scaffolds, are direct indicators of assembly fragmentation [3].
- Contig-to-Chromosome (CC) Ratio: A recently proposed metric that compensates for the flaws of N50. It is calculated as the number of contigs divided by the number of chromosome pairs. A lower ratio indicates a more contiguous assembly [13].
Completeness
Completeness assesses how much of the entire original genome sequence is present in the final assembly [11]. The goal is to minimize missing regions, whether they are genes or intergenic sequences.
- BUSCO (Benchmarking Universal Single-Copy Orthologs): BUSCO assesses completeness by searching for a set of highly conserved, single-copy orthologous genes that are expected to be present in a specific lineage. It reports the percentage of these genes found as "complete," "fragmented," "duplicated," or "missing." A BUSCO complete score above 95% is generally considered good [10] [3]. An elevated "duplicated" score can signal a problem with haplotig duplication or assembly errors [11].
- K-mer-based Completeness: This approach compares the k-mers (subsequences of length k) present in the raw sequencing reads to those found in the assembly. The proportion of read k-mers that are also present in the assembly is a direct measure of sequence completeness [13] [3]. Tools like Merqury are commonly used for this purpose [10].
- Mapping Rate: The percentage of original sequencing reads that successfully map back to the assembly can also indicate completeness, with a high mapping rate being desirable [3].
- Flow Cytometry Comparison: A pre-assembly method where the total length of the assembled genome is compared to an independent estimate of genome size obtained via flow cytometry [3].
Correctness
Correctness evaluates the accuracy of each base pair and the larger-scale structural integrity of the assembly [10] [3]. It is often considered the most challenging dimension to measure comprehensively.
- Base-Level Accuracy: This refers to the correctness of individual nucleotides.
- QV (Quality Value): A Phred-scaled score (e.g., QV 40 = 99.99% accuracy) that estimates the probability of an incorrect base call. It can be derived from k-mer comparisons using tools like Merqury [13].
- SNV/Indel Count: The number of single-nucleotide variants and insertions/deletions identified by mapping high-accuracy short reads or by comparing to a gold-standard reference [10] [14].
- Structural-Level Accuracy: This assesses whether the order, orientation, and copy number of sequences are correct.
- Misassembly Detection: Tools like QUAST can identify large-scale errors such as relocations, translocations, and inversions by aligning the assembly to a reference genome [15] [3].
- LTR Assembly Index (LAI): This metric evaluates the assembly quality of repetitive regions, specifically long terminal repeat (LTR) retrotransposons. A higher LAI (e.g., >10 for reference-quality) indicates that complex repetitive regions have been properly resolved [13] [3].
- Frameshift Analysis: The number of frameshifting indels within coding genes can be used as a proxy for assembly errors, as true frameshifts in conserved genes are rare [10].
The table below summarizes these key metrics for a quick reference.
Table 1: Summary of Core Genome Assembly Quality Metrics
| Dimension | Metric | Description | Target Value/Note |
|---|---|---|---|
| Contiguity | N50 / NG50 | Shortest contig length covering 50% of assembly/genome. | >1 Mb is often "good" [10]. |
| Number of Contigs | Total count of contiguous sequences. | Lower is better. | |
| CC Ratio | # Contigs / # Chromosome Pairs. | Compensates for N50 flaws; lower is better [13]. | |
| Completeness | BUSCO | % of conserved single-copy orthologs found. | >95% complete is "good" [10]. |
| K-mer Completeness | % of read k-mers found in the assembly. | Closer to 100% is better [3]. | |
| Mapping Rate | % of reads that map back to the assembly. | Closer to 100% is better [3]. | |
| Correctness | QV (Quality Value) | Phred-scaled base-level accuracy. | QV40 = 99.99% accuracy; higher is better [13]. |
| LAI (LTR Assembly Index) | Completeness of LTR retrotransposon assembly. | >10 for reference-quality [13]. | |
| # of Misassemblies | Large-scale errors (inversions, translocations). | Identified by QUAST; lower is better [15]. |
Experimental Protocols for Assessing Correctness
While contiguity and completeness can be assessed directly from the assembly and gene sets, evaluating correctness often requires more complex, orthogonal data and methodologies [10]. The following are established protocols for this purpose.
K-mer Spectrum Analysis with Merqury
Objective: To assess base-level accuracy (QV) and completeness without a reference genome. Data Required: Short-read Illumina data from the same individual. Workflow:
- K-mer Counting: Use a k-mer counter (e.g., Meryl) to build a database of all unique k-mers from the short-read data. This represents the "truth" set.
- Assembly K-mer Extraction: Extract all k-mers from the genome assembly.
- Comparison with Merqury: Run Merqury with the short-read k-mer database and the assembly.
- Analysis: Merqury outputs:
- QV Score: A consensus quality value for the entire assembly.
- K-mer Spectra Plot: A visualization showing k-mer multiplicity, which helps identify haplotypic duplications, collapsed repeats, and overall completeness.
- Error Tracks: Files that can be loaded into a genome browser (e.g., IGV) to visually inspect potential errors [10] [13].
Assessing Frameshifts with Transcriptome Data
Objective: To identify frameshifting indels in coding genes, which are often assembly errors. Data Required: High-quality transcript annotations or full-length RNA sequencing data (e.g., from PacBio Iso-Seq) from the same or a closely related sample [10]. Workflow:
- Alignment: Map the transcript sequences to the genome assembly.
- ORF Prediction: Identify open reading frames (ORFs) in the aligned transcripts.
- Frameshift Detection: Scan the alignments for indels that are not multiples of three, which disrupt the reading frame.
- Validation: Manually inspect frameshifts in a genomic context, as some may represent real biological variation. A high frequency of frameshifts in conserved genes strongly indicates assembly errors.
Validation in High-Confidence Regions
Objective: To measure assembly accuracy against a defined "gold standard" set of genomic regions. Data Required: A high-quality reference genome for the same species (but a different individual) and short-read data for the assembled sample [10]. Workflow:
- Define High-Confidence Regions: Map the short-read data from the assembled sample to the reference genome. Exclude regions with abnormal coverage, high variant density, or low mapping quality. The remaining regions are considered high-confidence.
- Align Assembly to Reference: Align the new assembly to the reference genome.
- Calculate Concordance: Measure the concordance (e.g., identity percentage, number of discrepancies) only within the high-confidence regions. This provides a realistic measure of accuracy in regions where the reference is reliable for the sample [10].
Polishing for Near-Perfect Accuracy
Objective: To correct residual errors in a long-read assembly, achieving accuracy suitable for outbreak investigation or high-resolution genomics. Data Required: A long-read (e.g., ONT) assembly and the original long reads, plus high-accuracy short reads (e.g., Illumina) from the same isolate. Experimental Insight: A 2024 benchmarking study on Salmonella outbreak isolates found that near-perfect accuracy (99.9999%) was only achieved by pipelines combining both long- and short-read polishing [14]. Recommended Workflow:
- Initial Assembly: Assemble with Flye.
- Long-Read Polishing: Polish the assembly with Medaka, which was found to be more accurate and efficient than Racon [14].
- Short-Read Polishing: Perform a subsequent round of polishing with a short-read polisher like NextPolish, Pilon, or POLCA. The study showed that Medaka followed by NextPolish was a top-performing combination [14]. Critical Note: The order of operations is crucial. Using a less accurate tool after a more accurate one can re-introduce errors [14].
The logical flow of a comprehensive assembly evaluation, integrating the "3 Cs" and various data types, can be visualized as follows:
Figure 1: A holistic workflow for genome assembly evaluation, integrating the three core dimensions (the "3 Cs") and their associated data requirements.
A Toolkit for the Assembly Scientist
Successful genome assembly and evaluation rely on a suite of bioinformatics tools and reagents. The following table details key solutions and their functions in the evaluation process.
Table 2: Essential Research Reagent Solutions for Genome Assembly Evaluation
| Category | Tool / Reagent | Primary Function in Evaluation |
|---|---|---|
| Quality Assessment Suites | QUAST [15] [3] | Comprehensive quality assessment with/without a reference; reports contiguity metrics and misassemblies. |
| GenomeQC [3] | Interactive web framework for comparing assemblies and benchmarking against gold standards. | |
| GAEP [3] | Comprehensive pipeline using NGS, long-read, and transcriptome data to assess all 3 Cs. | |
| Completeness Tools | BUSCO [3] [11] | Assesses gene space completeness using universal single-copy orthologs. |
| Merqury [10] [13] | Reference-free evaluation of quality (QV) and completeness using k-mers. | |
| Correctness & Polishing | Merqury / Yak [10] | K-mer-based base-level accuracy assessment. |
| Medaka [1] [14] | Long-read polisher that uses raw signal data to correct assembly errors. | |
| Racon [1] | A general long-read polisher. | |
| Pilon [1] | A general short-read polisher. | |
| NextPolish [14] | Short-read polisher identified as highly accurate in benchmarking. | |
| Structural Evaluation | QUAST [15] | Identifies large-scale misassemblies via reference alignment. |
| LAI Calculator [13] | Evaluates assembly quality in repetitive regions via LTR retrotransposon completeness. | |
| Orthogonal Data | PacBio Iso-Seq Data [10] | Full-length transcript sequences for validating gene models and detecting frameshifts. |
| Hi-C / Chicago Data [1] | Proximity-ligation data for scaffolding to chromosome scale and validating structural accuracy. | |
| Illumina Short Reads [10] [14] | High-accuracy reads for k-mer completeness analysis, polishing, and variant detection. |
Benchmarking genome assemblers requires a multi-faceted approach that moves beyond simplistic contiguity statistics. A rigorous evaluation must simultaneously consider contiguity, completeness, and correctness to paint a true picture of assembly quality. As demonstrated, this involves leveraging a suite of tools like QUAST, BUSCO, and Merqury, and employing orthogonal data through defined experimental protocols, such as k-mer analysis and hybrid polishing. The field is moving towards more holistic and biologically informed metrics, such as the LAI and CC ratio, to better capture the nuances of assembly quality. By adopting the comprehensive framework and metrics outlined in this guide, researchers can make informed decisions when selecting assemblers, confidently compare algorithmic performance, and ultimately generate genome assemblies that are not only well-assembled but also biologically accurate and truly useful for downstream scientific discovery.
Next-generation sequencing (NGS) has revolutionized genomics research, expanding our knowledge of genome structure, function, and dynamics [16]. The evolution from short-read sequencing to long-read sequencing technologies represents a paradigm shift in our ability to decipher genetic information with unprecedented completeness and accuracy. Short-read technologies, dominated by Illumina sequencing-by-synthesis approaches, have been the workhorse of genomics for over a decade, providing highly accurate (>99.9%) reads typically ranging from 50-300 base pairs [17] [18]. These technologies excel at identifying single nucleotide polymorphisms (SNPs) and small insertions/deletions efficiently and cost-effectively, making them ideal for applications like whole genome sequencing (WGS), whole exome sequencing (WES), and gene panel testing [17].
However, the limited read length of these platforms presents significant challenges for resolving complex genomic regions, including structural variations, large repetitive elements, and extreme GC-content regions [18]. Approximately 15% of the human genome remains inaccessible to short-read technologies, including centromeres, telomeres, and large segmental duplications—ironically, some of the most mutable regions of our genome [18]. These limitations have driven the development and refinement of long-read sequencing technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), which can generate reads tens to thousands of kilobases in length, enabling the complete assembly of genomes from telomere to telomere (T2T) [19] [18].
Table 1: Comparison of Major Sequencing Technologies
| Technology | Read Length | Accuracy | Primary Applications | Key Limitations |
|---|---|---|---|---|
| Illumina | 50-300 bp | >99.9% | WGS, WES, gene panels, SNP discovery | Limited resolution of repetitive regions, structural variants |
| PacBio HiFi | 10-25 kb | >99% | De novo assembly, structural variant detection, haplotype phasing | Higher cost per base, requires more DNA input |
| ONT | 10-60 kb (standard); up to >1 Mb (ultra-long) | 87-98% | Real-time sequencing, large structural variants, base modification detection | Higher raw error rate requires correction |
Technical Foundations of Short-Read and Long-Read Sequencing
Short-Read Sequencing Methodologies
Short read sequencing encompasses several technological approaches that determine nucleotide sequences in fragments typically ranging from 50-300 base pairs [17]. Sequencing by synthesis (SBS) platforms utilize polymerase enzymes to replicate single-stranded DNA fragments, employing either fluorescently-labeled nucleotides with reversible blockers that halt the reaction after each incorporation, or unmodified nucleotides that are introduced sequentially while detecting incorporation through released hydrogen ions and pyrophosphate [17]. The sequencing by binding (SBB) approach splits nucleotide incorporation into distinct steps: fluorescently-labeled nucleotides bind to the template without incorporation for signal detection, followed by washing and subsequent extension with unlabeled nucleotides [17]. Alternatively, sequencing by ligation (SBL) employs ligase enzymes instead of polymerase to join fluorescently-labeled nucleotide sequences to the template strand [17].
The exceptional accuracy of short-read technologies (>99.9%) makes them particularly suitable for variant calling applications where base-level precision is critical [18]. This high accuracy, combined with massive throughput capabilities (up to 3000 Gb per flow cell on Illumina NovaSeq 6000) and lower per-base cost, has cemented their position as the first choice for large-scale genomic studies requiring SNP identification and small indel detection [18] [16]. However, their fundamental limitation remains the inability to span repetitive regions or resolve complex structural variations that exceed their read length [18].
Long-Read Sequencing Platforms
Pacific Biosciences (PacBio) Technology
PacBio's single-molecule real-time (SMRT) sequencing utilizes a unique circular template design called a SMRTbell, comprised of a double-stranded DNA insert with single-stranded hairpin adapters on both ends [18]. This structure allows DNA polymerase to repeatedly traverse the circular template, enabling circular consensus sequencing (CCS) that generates highly accurate HiFi (High Fidelity) reads through multiple observations of each base [18]. The technology operates on a SMRT Cell containing millions of zero-mode waveguides (ZMWs)—nanophotonic structures that confine observation volumes to the single-molecule level, allowing real-time detection of nucleotide incorporation events [18] [16].
PacBio systems typically produce reads tens of kilobases in length, with recent advancements enabling read N50 lengths of 30-60 kb and maximum reads exceeding 200 kb [18]. The platform's unique capability to monitor the kinetics of nucleotide incorporation provides inherent access to epigenetic information, allowing direct detection of base modifications such as methylation without specialized sample preparation [17].
Oxford Nanopore Technologies (ONT)
ONT sequencing employs a fundamentally different approach based on the changes in electrical current as DNA molecules pass through protein nanopores embedded in a membrane [17] [18]. A constant voltage is applied across the membrane, and as negatively-charged single-stranded DNA molecules translocate through the nanopores, each nucleotide base causes characteristic disruptions in the ionic current that can be decoded to determine the DNA sequence [17]. This unique mechanism enables truly real-time sequencing and allows for the longest read lengths currently available, with standard protocols producing reads of 10-60 kb and ultra-long protocols generating reads exceeding 100 kb, with some reaching megabase lengths [18].
A distinctive advantage of the ONT platform is its capacity for direct RNA sequencing without reverse transcription, preserving native nucleotide modification information [17]. The technology's portability (particularly the MinION device) and rapidly improving throughput (up to 180 Gb per PromethION flow cell) have expanded sequencing applications to field-based and point-of-care scenarios [18].
Table 2: Performance Metrics of Long-Read Sequencing Platforms
| Parameter | PacBio (Sequel II) | ONT (PromethION) |
|---|---|---|
| Read Length N50 | 30-60 kb | 10-60 kb (standard); 100-200 kb (ultra-long) |
| Maximum Read Length | >200 kb | >1 Mb |
| Raw Read Accuracy | 87-92% (CLR); >99% (HiFi) | 87-98% |
| Throughput per Flow Cell | 50-100 Gb (CLR); 15-30 Gb (HiFi) | 50-100 Gb |
| Epigenetic Detection | Native detection of base modifications | Native detection of base modifications |
| RNA Sequencing | Requires cDNA synthesis | Direct RNA sequencing |
Benchmarking Genome Assemblers: From Short Reads to Complete Genomes
Performance Evaluation of Short-Read Simulators
Comprehensive benchmarking of computational tools is essential for reliable genomic analysis. A 2023 study evaluated six popular short-read simulators—ART, DWGSIM, InSilicoSeq, Mason, NEAT, and wgsim—assessing their ability to emulate characteristic features of empirical Illumina sequencing data, including genomic coverage, fragment length distributions, quality scores, systematic errors, and GC-coverage bias [20]. The research highlighted that these tools employ either pre-defined "basic" models or "advanced" parameterized custom models designed to mimic genomic characteristics of specific organisms, with significant variability in their ability to faithfully reproduce platform-specific artifacts and biological features [20].
Performance comparisons revealed substantial differences in how accurately these simulators replicated quality score distributions and GC-coverage biases present in real datasets [20]. Tools like InSilicoSeq offered extensive ranges of built-in platform-specific error models for common Illumina sequencers (HiSeq, NovaSeq, MiSeq), while others provided more flexibility for custom parameterization [20]. The study emphasized that careful simulator selection is crucial for generating meaningful synthetic datasets for pipeline benchmarking, particularly for non-model organisms lacking gold-standard reference datasets [20].
Long-Read Assembly Tool Benchmarking
As long-read technologies have matured, numerous assemblers have been developed to leverage their advantages. A comprehensive benchmarking of eleven long-read assemblers—Canu, Flye, HINGE, Miniasm, NECAT, NextDenovo, Raven, Shasta, SmartDenovo, wtdbg2 (Redbean), and Unicycler—using standardized computational resources revealed significant differences in performance [21]. Assemblers employing progressive error correction with consensus refinement, notably NextDenovo and NECAT, consistently generated near-complete, single-contig assemblies with low misassemblies and stable performance across preprocessing types [21]. Flye offered a strong balance of accuracy and contiguity, while Canu achieved high accuracy but produced more fragmented assemblies (3-5 contigs) with substantially longer runtimes [21].
Ultrafast tools like Miniasm and Shasta provided rapid draft assemblies but were highly dependent on preprocessing and required polishing to achieve completeness [21]. The study also demonstrated that preprocessing decisions significantly impact assembly quality, with filtering improving genome fraction and BUSCO completeness, trimming reducing low-quality artifacts, and correction benefiting overlap-layout-consensus (OLC)-based assemblers while occasionally increasing misassemblies in graph-based tools [21].
A separate 2025 benchmarking study of hybrid de novo assembly pipelines combining ONT long-reads with Illumina short-reads found that Flye outperformed all assemblers, particularly when using Ratatosk error-corrected long-reads [5]. Post-assembly polishing significantly improved accuracy and continuity, with two rounds of Racon (long-read-based polishing) followed by Pilon (short-read-based polishing) yielding optimal results [5]. This comprehensive evaluation highlighted that hybrid approaches effectively integrate the long-range continuity of ONT data with the base-level accuracy of Illumina reads, providing a balanced solution for high-quality genome assembly [5].
Telomere-to-Telomere Assembly: Case Studies and Breakthroughs
The Telomere-to-Telomere Assembly Paradigm
Telomere-to-telomere (T2T) assembly represents the ultimate goal of genome sequencing—complete, gap-free chromosome assemblies that include traditionally challenging regions such as centromeres, telomeres, and ribosomal DNA (rDNA) arrays [19] [22]. Long-read technologies have been instrumental in achieving this milestone, with T2T assemblies now completed for multiple species including human, banana, and hexaploid wheat [19] [22]. These complete assemblies reveal unprecedented insights into genome biology, enabling comprehensive characterization of previously inaccessible genomic features and their role in evolution, disease, and fundamental biological processes [19].
The power of T2T assemblies lies in their ability to resolve complex regions that have historically plagued genome projects. Centromeres, characterized by megabase-scale tandem repeats, are essential for chromosome segregation but were previously largely unassembled [19]. Telomeres, composed of repetitive sequences at chromosome ends, protect genomic integrity but vary substantially between species and even within individuals [23]. Ribosomal DNA clusters, comprised of highly similar tandemly repeated genes, challenge assembly algorithms due to their extensive homogeneity [22]. T2T assemblies now enable systematic study of these regions, revealing their architecture, variation, and functional significance.
Plant Genome T2T Assemblies
Banana Genome Assembly
A landmark 2021 study demonstrated the power of ONT long-read sequencing for plant genome assembly, generating a chromosome-scale assembly of banana (Musa acuminata) with five of eleven chromosomes entirely reconstructed in single contigs from telomere to telomere [22]. Using a single PromethION flowcell generating 93 Gb of sequence (177X coverage) with read N50 of 31.6 kb, the assembly achieved remarkable contiguity with the NECAT assembler, producing an assembly comprised of just 124 contigs with a cumulative size of 485 Mbp [22]. Validation using two independent Bionano optical maps (DLE-1 and BspQI enzymes) confirmed assembly accuracy, with only one small contig (380 kbp) flagged as conflictual [22].
This T2T assembly revealed, for the first time, the complete architecture of complex regions including centromeres and clusters of paralogous genes [22]. All eleven chromosome sequences harbored plant-specific telomeric repeats (T3AG3) at both ends, confirming complete assembly of chromosome termini [22]. The remaining gaps were primarily located in rDNA clusters (5S for chromosomes 1, 3, and 8; 45S for chromosome 10) and other tandem and inverted repeats, highlighting that even with long-read technologies, these extremely homogeneous repetitive regions remain challenging to resolve completely [22].
Hexaploid Wheat Genome Assembly
The recent CS-IAAS assembly of hexaploid bread wheat (Triticum aestivum L.) represents a monumental achievement in plant genomics, producing a complete T2T gap-free genome encompassing 14.51 billion base pairs with all 21 centromeres and 42 telomeres [19] [24]. This assembly utilized a sophisticated hybrid approach combining PacBio HiFi reads (3.8 Tb, ~250× coverage) with ONT ultra-long reads (>100 kbp, 1.8 Tb, ~120× coverage), supplemented with Hi-C, Illumina, and Bionano data [19]. The development of a semi-automated pipeline for assembling reference sequence of T2T (SPART) enabled the integration of these complementary technologies, leveraging the precision of HiFi sequencing and the exceptional contiguity of ONT ultra-long reads [19].
The resulting assembly demonstrated dramatic improvements over previous versions, with contig N50 increasing from 0.35 Mbp in CS RefSeq v2.1 to 723.78 Mbp in CS-IAAS—a 206,694% improvement—while completely eliminating all 183,603 gaps present in the previous assembly [19] [24]. This comprehensive genome enabled the identification of 565.66 Mbp of new sequences, including centromeric satellites (16.05%), transposable elements (68.66%), rDNA arrays (0.75%), and other previously inaccessible regions [19]. The complete assembly facilitated unprecedented analysis of genome-wide rearrangements, centromeric elements, transposable element expansion, and segmental duplications during tetraploidization and hexaploidization, providing comprehensive understanding of wheat subgenome evolution [19].
Computational Methods for Telomere Analysis
The expansion of long-read sequencing has driven development of specialized computational methods for analyzing telomeres. Traditional experimental methods for telomere length measurement, such as terminal restriction fragment (TRF) assay and quantitative fluorescence in situ hybridization (Q-FISH), face limitations including high DNA requirements, labor intensity, and challenges in scaling for high-throughput studies [23]. Computational methods like TelSeq, Computel, and TelomereHunter have been developed to estimate telomere length from short-read sequencing data by quantifying telomere repeat abundance, but these methods show only moderate correlation with experimental techniques (Spearman's ρ = 0.55 between K-seek and TRF in A. thaliana) and remain susceptible to biases from library preparation and PCR amplification [23].
The Topsicle method, introduced in 2025, represents a significant advance by estimating telomere length from whole-genome long-read sequencing data using k-mer and change-point detection analysis [23]. This approach leverages the ability of long reads to span entire telomere-subtelomere junctions, enabling precise determination of the boundary position and subsequent length calculation [23]. Simulations demonstrate robustness to sequencing errors and coverage variations, with application to plant and human cancer cells showing high accuracy comparable to direct telomere length measurements [23]. This tool is particularly valuable because it accommodates the diverse telomere repeat motifs found across different species, unlike previous methods optimized primarily for the human TTAGGG motif [23].
Experimental Design and Methodologies for Genome Assembly
Recommended Workflows for T2T Assembly
Based on benchmarking studies and successful T2T assemblies, optimal genome assembly workflows integrate multiple technologies and analysis steps. For long-read-only assembly, the recommended workflow includes: (1) high-molecular-weight DNA extraction using protocols optimized for long fragments; (2) sequencing with either PacBio HiFi or ONT ultra-long protocols to achieve sufficient coverage (>50X); (3) assembly with assemblers like Flye, NECAT, or NextDenovo that have demonstrated strong performance in benchmarks; (4) iterative polishing with long-read data using tools like Racon or Medaka; and (5) optional short-read polishing with tools like Pilon for maximum base-level accuracy [21] [22] [5].
For hybrid assembly approaches that combine long-read and short-read technologies: (1) sequence with both ONT (for contiguity) and Illumina (for accuracy) platforms; (2) perform pre-assembly error correction of long reads using tools like Ratatosk with short-read data; (3) assemble with hybrid-aware assemblers; (4) conduct multiple rounds of polishing with both long-read and short-read polishers; and (5) validate assembly quality using multiple metrics including BUSCO completeness, Merqury QV scores, and optical mapping [5]. Chromosome-scale scaffolding can be achieved through additional Hi-C or optical mapping data, with the Dovetail Omni-C and Bionano systems providing complementary approaches for validating and improving scaffold arrangements [19] [22].
Diagram 1: Complete T2T Genome Assembly Workflow. This workflow integrates laboratory and computational phases, highlighting the multi-stage process required for successful telomere-to-telomere assembly.
Essential Research Reagents and Tools
Table 3: Essential Research Reagents and Computational Tools for Genome Assembly
| Category | Specific Tools/Reagents | Function | Application Context |
|---|---|---|---|
| DNA Extraction | Circulomics SRE XL kit | Removal of short DNA fragments | HMW DNA preparation for long-read sequencing |
| Sequencing Kits | PacBio SMRTbell Express Template Prep Kit 2.0 | Library preparation for PacBio sequencing | HiFi read generation |
| ONT Ligation Sequencing Kit (SQK-LSK109) | Library preparation for Nanopore sequencing | Standard long-read generation | |
| Assembly Software | Flye, NECAT, NextDenovo | De novo genome assembly from long reads | Production of contiguous assemblies |
| Polishing Tools | Racon, Medaka | Long-read-based consensus polishing | Error correction after assembly |
| Pilon | Short-read-based polishing | Final base-level accuracy improvement | |
| Validation Tools | BUSCO, Merqury | Assembly completeness and quality assessment | Benchmarking assembly quality |
| Bionano Solve | Optical mapping analysis | Scaffold validation and conflict resolution |
The evolution from short-read to long-read sequencing technologies has fundamentally transformed genomics, enabling complete telomere-to-telomere assemblies that reveal previously inaccessible regions of genomes [19] [18]. Benchmarking studies have demonstrated that both PacBio HiFi and ONT ultra-long reads can produce exceptionally contiguous assemblies, with assembler selection significantly impacting outcomes [21] [5]. The development of specialized computational methods like Topsicle for telomere analysis further enhances the utility of long-read data for investigating fundamental biological questions [23].
As these technologies continue to mature, several trends are shaping the future of genome sequencing and assembly. Continuous improvements in read length and accuracy are making T2T assemblies more routine and accessible [18] [16]. The integration of multiple complementary technologies—PacBio for accuracy, ONT for length, Hi-C for scaffolding, and optical mapping for validation—represents the current state-of-the-art for complex genomes [19] [22]. Computational methods are advancing rapidly to leverage these data, with specialized assemblers and polishers improving both contiguity and accuracy [21] [5].
For researchers and drug development professionals, these advances translate to more comprehensive understanding of genetic variation and its functional consequences. Complete genome assemblies enable systematic study of previously neglected repetitive regions, revealing their roles in disease, evolution, and genomic stability [23] [19]. As T2T assemblies become more commonplace, we anticipate discoveries linking variation in complex genomic regions to phenotypic outcomes, potentially unlocking new therapeutic targets and diagnostic approaches [18]. The ongoing evolution of sequencing technologies and computational methods promises to further democratize access to complete genome sequencing, ultimately advancing personalized medicine and fundamental biological discovery.
Assembly in Action: Strategies, Tools, and Pipelines for Robust Genome Reconstruction
Genome assembly is a foundational step in genomics, critically influencing downstream applications such as functional annotation, comparative genomics, and variant discovery [21]. The overarching goal of any genome assembler is to reconstruct the complete genome in the fewest possible contiguous pieces (contigs/scaffolds) with the highest base accuracy, while minimizing computational resource consumption [25]. Achieving these "1-2-3 goals" is challenging due to pervasive repetitive sequences and sequencing errors. The human genome, for instance, is estimated to be 66–69% repetitive, making the resolution of these regions paramount for a successful assembly [26]. Over the years, distinct algorithmic paradigms have been developed to tackle these challenges, primarily falling into three categories: Overlap-Layout-Consensus (OLC), graph-based (primarily de Bruijn graphs), and hybrid approaches. This guide provides an objective comparison of these paradigms, drawing on recent benchmarking studies to evaluate their performance, optimal use cases, and computational requirements.
Assembly Paradigms and Core Algorithms
Overlap-Layout-Consensus (OLC)
The OLC paradigm, a classical approach adapted for long reads, involves three main steps. First, the Overlap step performs an all-versus-all pairwise comparison of reads to find overlaps. Second, the Layout step uses these overlaps to construct a graph and determine the order and orientation of reads. Finally, the Consensus step derives the final sequence by determining the most likely nucleotide at each position from the multiple alignments [25] [26]. This paradigm is naturally suited for long, error-prone reads because it can tolerate a higher error rate during the initial overlap detection. Modern OLC assemblers have introduced significant optimizations to handle the computational burden of all-versus-all read comparison. For example, Flye clusters long reads from the same genomic locus to reduce comparisons [26], Redbean segments reads to speed up alignment [26], and Shasta uses run-length encoding to compress homopolymers, mitigating a common error type in Oxford Nanopore Technologies (ONT) data [26].
Graph-Based (de Bruijn Graph)
In contrast to OLC, the de Bruijn graph approach breaks all reads into short, fixed-length subsequences called k-mers. The assembly is then reconstructed by finding a path that traverses every k-mer exactly once (an Eulerian path) [25]. This method is highly efficient for large volumes of accurate, short-read data because it avoids the computationally expensive all-versus-all read comparison. However, the process of splitting reads into k-mers can cause a loss of long-range information, making it less ideal for resolving long repeats when using only short reads. While traditionally used for short reads, innovations like the one in GoldRush demonstrate how de Bruijn graph principles can be adapted for long-read assembly by using a dynamic, probabilistic multi-index Bloom filter data structure to achieve linear time complexity and a dramatically reduced memory footprint [26].
Hybrid Assembly
Hybrid assemblers integrate data from both long-read (e.g., ONT, PacBio) and short-read (e.g., Illumina) technologies to leverage their complementary strengths. The long reads provide the contiguity, while the highly accurate short reads correct base-level errors. Strategies vary; some tools follow a "long-read-first" approach where the assembly is primarily built from long reads and then polished with short reads [27] [14]. Others, like WENGAN, implement a "short-read-first" strategy. WENGAN starts by building a de Bruijn graph from short reads, then uses synthetic paired reads derived from long reads to build a "synthetic scaffolding graph" (SSG), which is used to order contigs and fill gaps with long-read consensus sequences [25]. This approach avoids the all-versus-all long-read comparison and efficiently integrates data types from the start.
Table 1: Overview of Genome Assembly Paradigms and Representative Tools
| Assembly Paradigm | Core Principle | Representative Tools | Ideal Sequencing Data |
|---|---|---|---|
| Overlap-Layout-Consensus (OLC) | Finds overlaps between long reads to build a layout and consensus sequence. | Flye, Canu, Shasta, Redbean, NECAT, NextDenovo [21] [26] | Long-reads only (ONT, PacBio CLR) |
| de Bruijn Graph | Splits reads into k-mers and reconstructs the genome via Eulerian paths. | MEGAHIT, GoldRush (adapted) [25] [26] | Short-reads only (Illumina) |
| Hybrid | Combines long and short reads for scaffolding and error correction. | Unicycler, MaSuRCA, SPAdes, WENGAN [27] [25] [28] | Long-reads + Short-reads |
Benchmarking Performance and Computational Efficiency
Recent large-scale benchmarks provide critical insights into the performance of these paradigms. A 2024 study evaluating polishing tools highlighted that near-perfect accuracy for bacterial genomes (99.9999%) is only achievable with pipelines that combine both long-read assembly and short-read polishing [14].
A comprehensive benchmark of eleven long-read assemblers on microbial genomes found that assemblers employing progressive error correction, notably NextDenovo and NECAT, consistently generated near-complete, single-contig assemblies with low misassemblies. Flye offered a strong balance of accuracy and contiguity, while Canu achieved high accuracy at the cost of more fragmented assemblies (3–5 contigs) and the longest runtimes. Ultrafast tools like Miniasm and Shasta provided rapid drafts but were highly dependent on pre-processing and required polishing for completeness [21].
For the demanding task of human genome assembly, a 2025 benchmarking study found that Flye outperformed all other assemblers, particularly when long reads were error-corrected with Ratatosk prior to assembly [27] [5]. The study also confirmed that polishing, especially two rounds of Racon (long-read) followed by Pilon (short-read), consistently improved both assembly accuracy and continuity [27].
In terms of computational resource usage, a notable departure from the OLC paradigm is GoldRush. When assembling human genomes, GoldRush achieved contiguity (NG50 25.3–32.6 Mbp) comparable to Shasta and Flye, but did so using at most 54.5 GB of RAM. This is a fraction of the resources required by Flye (329.3–502.4 GB) and Shasta (884.8–1009.2 GB), demonstrating the potential for new algorithms to drastically improve scalability [26].
Table 2: Performance and Resource Usage of Select Assemblers from Benchmarking Studies
| Assembler | Paradigm | Contiguity (Human NG50) | Key Strengths | Computational Cost | Best Use Cases |
|---|---|---|---|---|---|
| Flye [27] [26] | OLC | 26.6 - 38.8 Mbp | High accuracy & contiguity balance; top performer in human assembly [27]. | High RAM (329-502 GB), long runtime (>33.7h) [26]. | Standard for large, complex genomes. |
| NextDenovo [21] | OLC | N/A (Microbial) | Near-complete microbial assemblies; low misassemblies; stable performance [21]. | N/A | Prokaryotic genomics; high-contiguity microbial assemblies. |
| Shasta [21] [26] | OLC | 29.7 - 39.6 Mbp | Ultrafast assembly; suitable for haploid assembly [26]. | Very High RAM (885-1009 GB) [26]. | Rapid draft assembly of large genomes. |
| GoldRush [26] | Graph-based | 25.3 - 32.6 Mbp | Linear time complexity; low RAM (<54.5 GB); correct assemblies [26]. | Low RAM, fast (<20.8h for human) [26]. | Resource-constrained environments; large-scale projects. |
| Unicycler [28] | Hybrid | N/A (Bacterial) | Superior for bacterial genomes; produces contiguous, circular assemblies [28]. | N/A | Bacterial pathogen genomics; complete circular genomes. |
| WENGAN [25] | Hybrid | 17.24 - 80.64 Mbp | High contiguity & quality; efficient; effective at low long-read coverage [25]. | Low computational cost (187-1200 CPU hours) [25]. | Human and large eukaryotic genomes. |
Experimental Protocols in Benchmarking Studies
To ensure the reproducibility of assembly benchmarks, studies follow rigorous, standardized protocols. Below is a detailed methodology common to recent comprehensive evaluations.
Data Acquisition and Pre-processing
Benchmarks typically use well-characterized reference samples, such as the HG002 (NA24385) human sample from the Genome in a Bottle (GIAB) consortium [27] [5]. Data includes both long reads (e.g., ~47x coverage from ONT PromethION) and short reads (e.g., ~35x coverage from Illumina NovaSeq 6000) [5]. Pre-processing is a critical step that can markedly affect assembly quality. Common procedures include:
- Filtering: Removes low-quality reads to improve genome fraction and BUSCO completeness [21].
- Trimming: Trims adapters and low-quality bases to reduce artifacts [21].
- Error Correction: Correcting long reads before assembly (e.g., with Ratatosk) can benefit OLC-based assemblers, though it may occasionally increase misassemblies in graph-based tools [21] [27].
Assembly Execution and Polishing
The selected assemblers are run on the pre-processed reads using standardized computational resources. A key finding across studies is that polishing is essential for achieving high accuracy with long-read assemblies [27] [14]. The optimal polishing strategy identified in multiple benchmarks is:
- Long-read polishing: First, perform one or more rounds of polishing using long reads themselves, with tools like Medaka or Racon. Medaka has been shown to be more accurate and efficient than Racon [14].
- Short-read polishing: Follow with one or more rounds of polishing using high-accuracy short reads. Tools like NextPolish, Pilon, Polypolish, and POLCA perform similarly, with NextPolish showing the highest accuracy in some studies [14]. The order matters—using less accurate tools after more accurate ones can introduce errors [14].
Quality Assessment and Metrics
Assemblies are evaluated using a suite of complementary metrics to assess different aspects of quality:
- Contiguity: Assessed via N50/NG50 (the contig length at which 50% of the genome is assembled) and contig count [21] [26].
- Completeness: Measured with BUSCO (Benchmarking Universal Single-Copy Orthologs), which quantifies the presence of expected evolutionarily conserved genes [21] [27].
- Base-level Accuracy: Evaluated using Merqury (which computes consensus quality values, QV) and variant calling [27] [26].
- Structural Accuracy: Tools like QUAST report misassemblies and the NGA50, which is the NG50 after breaking contigs at misassembly sites [27] [26].
- Gene-Level Accuracy: The
asmgeneutility in minimap2 can be used to assess the accuracy of gene regions [26].
Diagram Title: Standard Workflow for Benchmarking Genome Assemblers
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Bioinformatics Tools and Resources for Genome Assembly and Evaluation
| Tool / Resource | Category | Primary Function | Citation |
|---|---|---|---|
| Flye | Assembler | OLC-based long-read assembly for large genomes. | [21] [27] [26] |
| Unicycler | Assembler | Hybrid assembler optimized for bacterial genomes. | [28] |
| Medaka | Polishing | Long-read polisher for ONT data; accurate and efficient. | [14] |
| Racon | Polishing | Consensus-based polisher for long reads. | [27] [14] |
| NextPolish | Polishing | Short-read polisher; high accuracy. | [14] |
| Pilon | Polishing | Short-read polisher for improving draft assemblies. | [27] |
| QUAST | Evaluation | Quality Assessment Tool for Genome Assemblies. | [27] [26] |
| BUSCO | Evaluation | Assesses assembly completeness based on conserved genes. | [21] [27] |
| Merqury | Evaluation | Evaluates consensus quality (QV) and assembly accuracy. | [27] [26] |
| HG002/NA24385 | Reference | GIAB reference material for benchmarking human assemblies. | [27] [5] |
The evidence from recent benchmarking studies indicates that there is no single "universally optimal" assembler; the choice depends on the organism, data type, and computational resources [21].
- For large, complex eukaryotic genomes (e.g., human): Flye is a robust, high-performing choice, especially when combined with Ratatosk pre-correction and a Racon-Medaka-Pilon polishing strategy [27]. For projects with limited computational resources, GoldRush offers a compelling alternative with linear time complexity and minimal RAM usage without sacrificing contiguity [26].
- For bacterial genomes: Unicycler is the superior approach, reliably producing contiguous and often circularized genomes from hybrid data [28].
- To achieve the highest possible accuracy: A hybrid approach that combines long-read assembly with both long- and short-read polishing is essential. This is particularly critical for applications like outbreak source tracking, where near-perfect accuracy is required [14].
Ultimately, assembler choice and pre-processing methods jointly determine the accuracy, contiguity, and computational efficiency of the final genome assembly, and should be carefully considered in the context of the specific research goals [21].
Long-read sequencing technologies have revolutionized genomics by enabling the assembly of complex genomic regions that were previously intractable. The choice of de novo assembler is a critical decision that directly impacts the contiguity, accuracy, and completeness of the resulting genome. This comparison guide objectively evaluates the performance of four prominent long-read assemblers—Flye, NextDenovo, Canu, and Shasta—within the established context of genome assembler benchmarking research. We synthesize findings from recent, rigorous studies to provide researchers and bioinformaticians with a data-driven foundation for selecting appropriate tools for their projects.
Performance Benchmarks and Comparative Analysis
Comprehensive benchmarking studies provide critical insights into the strengths and weaknesses of each assembler. Performance varies based on the genome being assembled, read characteristics, and computational resources.
Table 1: Summary of Assembler Performance Based on Recent Benchmarking Studies
| Assembler | Assembly Strategy | Contiguity (N50) | Completeness (BUSCO) | Base Accuracy | Computational Speed | Key Strengths |
|---|---|---|---|---|---|---|
| Flye | Assembly Then Correction (ATC) | Consistently High [27] | High [27] | High (especially with polishing) [27] | Moderate to Fast [21] | Excellent balance of accuracy and contiguity; robust performance [27] [21] |
| NextDenovo | Correction Then Assembly (CTA) | Very High [29] [21] | Near-Complete [21] | Very High (>99%) [29] | Very Fast [29] [21] | High speed and accuracy; efficient for noisy reads and large genomes [29] |
| Canu | Correction Then Assembly (CTA) | High (can be fragmented) [21] | High [21] | High [21] | Slow [29] [21] | High accuracy; thorough error correction [21] |
| Shasta | Assembly Then Correction (ATC) | Variable [21] | Requires Polishing [21] | Requires Polishing [21] | Ultrafast [21] | Extremely rapid assembly; good for initial drafts [21] |
Table 2: Performance on Human and Microbial Genomes
| Assembler | Human Genome (HG002) Performance [27] | Microbial Genome Performance [21] |
|---|---|---|
| Flye | Top performer, especially with error-corrected reads and polishing [27]. | Strong balance of accuracy and contiguity; sensitive to input read quality [21]. |
| NextDenovo | Validated for population-scale human assembly; accurate segmental duplication resolution [29]. | Consistently generates near-complete, single-contig assemblies with low misassemblies [21]. |
| Canu | Not the top performer in recent human benchmarks [27]. | High accuracy but often produces 3–5 contigs; longest runtimes [21]. |
| Shasta | Performance not specifically highlighted in the human benchmark [27]. | Provides rapid drafts but is highly dependent on pre-processing; requires polishing for completeness [21]. |
Detailed Methodologies of Key Experiments
The performance data presented above is derived from standardized benchmarking protocols. Understanding these methodologies is crucial for interpreting the results and designing your own experiments.
Benchmarking of Hybrid De Novo Assembly for Human Genomes
A 2025 study provided a comprehensive evaluation of assemblers using the HG002 human reference material [27].
- Sequencing Data: The benchmark utilized whole-genome sequencing data from Oxford Nanopore Technologies (ONT) for long reads and Illumina for short-read data [27].
- Assemblers & Polishing Tested: The study evaluated 11 pipelines, including Flye, NextDenovo, Canu, and Shasta. These were combined with four different polishing schemes (e.g., Racon, Pilon). The best-performing pipeline was further validated on non-reference human and non-human samples [27].
- Evaluation Metrics: Assembly quality was assessed using QUAST (for contiguity and misassembly metrics), BUSCO (for completeness based on universal single-copy orthologs), and Merqury (for base-level accuracy). Computational costs were also analyzed [27].
- Key Workflow: The optimal pipeline identified involved error correction of long reads with Ratatosk, assembly with Flye, and polishing with two rounds of Racon followed by Pilon [27].
Evaluation of Error Correction and Assembly for Noisy Long Reads
The development and assessment of NextDenovo involved rigorous benchmarking against other CTA assemblers [29].
- Data Sets: Both simulated data and real ONT "ultra-long" read data from the CHM13 human genome were used. The real data had an average read length of 91.21 kb [29].
- Compared Tools: NextDenovo was benchmarked against Consent, Canu, and Necat for its error correction module, and against a wider range of assemblers for the final assembly [29].
- Evaluation Metrics: For error correction, the study measured correction speed, the proportion of data successfully corrected, the final error rate of corrected reads, and the chimeric read rate. For the final assembly, standard metrics like contiguity and completeness were used [29].
Benchmarking Long-Read Assemblers for Prokaryotic Genomics
A study focused on microbial genomics benchmarked eleven long-read assemblers using standardized computational resources [21].
- Data and Pre-processing: Assemblies were evaluated across different pre-processing methods (raw, filtered, trimmed, and corrected reads) to assess the impact of data quality [21].
- Standardized Resources: All assemblers were run using the same computational environment to ensure a fair comparison of runtime and resource consumption [21].
- Evaluation Metrics: Assemblies were judged on runtime, standard contiguity metrics (N50, total length, contig count), GC content deviation, and completeness assessed via Benchmarking Universal Single-Copy Orthologs (BUSCO) [21].
Workflow Visualization of Assembly Strategies
Long-read assemblers primarily employ one of two core strategies. The diagram below illustrates the steps and logical relationships involved in the "Correction Then Assembly" (CTA) and "Assembly Then Correction" (ATC) approaches.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key bioinformatics tools and resources essential for conducting a robust assembly benchmark or performing genome assembly, as cited in the featured experiments.
Table 3: Key Research Reagent Solutions for Genome Assembly and Benchmarking
| Tool / Resource | Function | Relevance in Experiments |
|---|---|---|
| QUAST | Quality Assessment Tool for Genome Assemblies | Used to evaluate contiguity statistics (N50, contig count) and identify potential misassemblies [27]. |
| BUSCO | Benchmarking Universal Single-Copy Orthologs | Assesses assembly completeness by searching for a set of evolutionarily conserved genes expected to be present in single copy [27] [21]. |
| Merqury | Reference-free assembly evaluation suite | Evaluates base-level accuracy and quality value (QV) scores of an assembly using k-mer spectra [27]. |
| Racon | Ultrafast consensus module for genome assembly | Used as a polishing tool to correct errors in draft assemblies, often applied multiple times for best results [27]. |
| Pilon | Integrated tool for variant calling and assembly improvement | Used after Racon for final polishing, often leveraging Illumina short-read data for higher base accuracy [27]. |
| Ratatosk | Long-read error correction tool | Used to pre-correct ONT long reads before assembly with Flye, leading to superior performance [27]. |
| Oxford Nanopore (ONT) Data | Source of long-read sequencing data | Provides long reads (often >100 kb) crucial for spanning repeats; characterized by higher noise than other technologies [27] [29]. |
| Illumina Data | Source of short-read sequencing data | Used for polishing assemblies to achieve high base accuracy and for hybrid assembly approaches [27]. |
De novo genome assembly is a foundational process in genomics, enabling the decoding of genetic information for non-model organisms and providing critical insights into genome structure, evolution, and function [30]. The complete workflow, from raw sequencing reads to chromosome-scale assemblies, has been revolutionized by long-read sequencing technologies and proximity-ligation methods like Hi-C. However, constructing an optimal genome assembly requires careful selection of tools and strategies at each step, as the synergistic combination of sequencing technologies and specific software programs critically impacts the final output quality [31]. This guide provides an objective comparison of performance across assembly, polishing, and scaffolding tools, supported by experimental data from recent benchmarking studies, to inform researchers designing genome assembly pipelines.
Sequencing Platforms and Data Considerations
The choice of sequencing technology fundamentally influences assembly quality by determining the initial read characteristics. Second-generation sequencing (SGS) platforms like Illumina NovaSeq 6000 and MGI DNBSEQ-T7 provide highly accurate short reads (up to 99.5% accuracy) but struggle with repetitive regions and heterozygosity, often resulting in fragmented assemblies [31]. Third-generation sequencing (TGS) platforms, including Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), address these limitations by producing long reads spanning repetitive regions, despite having higher error rates (5-20%) [31].
Research indicates that input data with longer read lengths generally produce more contiguous and complete assemblies than shorter read length data with higher coverage [1]. A comprehensive study assembling yeast genomes found that ONT reads with R7.3 flow cells generated more continuous assemblies than those from PacBio Sequel, despite homopolymer-based errors and chimeric contigs [31]. For optimal results, more than 30× nanopore data is recommended, with quality highly dependent on subsequent polishing using complementary data [30].
Table 1: Sequencing Platform Characteristics
| Platform | Read Length | Error Rate | Error Profile | Best Use Case |
|---|---|---|---|---|
| Illumina | Short (150-300 bp) | <0.1% [1] | Substitution errors [31] | Polishing, variant calling |
| PacBio SMRT | Long (10-20 kb) | <1% [1] | Random errors | De novo assembly, repetitive regions |
| ONT | Long (up to hundreds of kb) | <5% [1] | Indel errors [31] | Structural variants, base modification |
Genome Assemblers Performance Comparison
Assembly Algorithms and Strategic Selection
De novo assemblers employ different algorithms with distinct advantages. Canu performs extensive error correction and trimming using overlap-consensus methods based on string graph theory, making it suitable for highly accurate assemblies despite substantial computational requirements [1] [31]. Flye identifies "disjointigs" and resolves repeat graphs using a generalized Bruijn graph approach, balancing contiguity and computational efficiency [1] [31]. WTDBG2 (now RedBean) uses a fuzzy DeBruijn algorithm optimized for speed with minimal computational resources [1] [31]. NECAT employs a progressive two-step error correction specifically designed for Nanopore raw reads [30].
The performance of these tools varies significantly based on coverage depth, with studies showing coverage depth has a substantial effect on final genome quality [30]. For low coverages (<16×), SPAdes has demonstrated superior N50 values compared to other assemblers in benchmarking studies [32].
Comparative Performance Metrics
A systematic evaluation of nine de novo assemblers for ONT data across different coverage depths revealed dramatic variations in contiguity among tools [30]. Another study benchmarking seven popular assemblers found they could be grouped into two classes based on N50 values, with SPAdes, Velvet, Discovar, MaSuRCA, and Newbler producing higher average N50 values than SOAP2 and ABySS across different coverage values [32].
Hybrid assemblers like MaSuRCA extend accurate SGS reads to their maximum unique length, connecting these "super-reads" using long TGS reads, which can mitigate the high error rates of TGS platforms [31]. For human genome assembly, a comprehensive benchmark of 11 pipelines found Flye outperformed all assemblers, particularly when using Ratatosk error-corrected long reads [33].
Table 2: Genome Assembler Performance Comparison
| Assembler | Algorithm Type | Key Characteristics | Optimal Coverage | Computational Demand |
|---|---|---|---|---|
| Canu | Overlap-Layout-Consensus | Multiple error correction rounds; high accuracy [31] | High (>50×) | High [30] [31] |
| Flye | Generalized Bruijn Graph | Efficient repeat resolution; good contiguity [31] | Moderate (30-50×) | Moderate [33] |
| WTDBG2 | Fuzzy DeBruijn Graph | Fast assembly with minimal resources [1] | Moderate (30-50×) | Low [31] |
| NECAT | Progressive correction | Optimized for Nanopore reads [30] | Moderate (30-50×) | Moderate |
| MaSuRCA | Hybrid | "Super-reads" from SGS with TGS links [31] | Varies by data type | Moderate |
Polishing Strategies for Accuracy Improvement
Polishing strategies are essential for correcting errors in initial assemblies. Polishers fall into two categories: "sequencer-bound" tools like Nanopolish and Medaka that utilize raw signal information, and "general" polishers like Racon and Pilon applicable to any sequencing platform [1]. Research indicates that iterative polishing progressively improves assembly accuracy, making previously unmappable reads available for subsequent rounds [1].
The most effective approach often combines multiple polishers. In benchmarking studies, the optimal polishing strategy involved two rounds of Racon followed by Pilon polishing [33]. Another study found that a combined Racon/Medaka/Pilon approach produced the most accurate final genome assembly [1]. For ONT data specifically, more than 30× coverage is recommended, with quality highly dependent on polishing using next-generation sequencing data [30].
Hi-C Scaffolding Tools Comparison
Scaffolding Algorithms and Approaches
Hi-C technology leverages proximity-based ligation and massively parallel sequencing to identify chromatin interactions across the entire genome, enabling contig grouping, ordering, and orientation into chromosome-scale assemblies [34]. The underlying principle is that Hi-C signal strength is stronger within chromosomes than between them, and within chromosomal regions, signals are more robust between physically proximate contigs [35].
Multiple Hi-C scaffolding tools have been developed with different computational strategies:
- YaHS creates a contact matrix by cutting contigs at essential breakpoints, builds a graph, and produces scaffolded outcomes after refinements [35].
- SALSA2 builds a hybrid graph combining information from ambiguous edges derived from the GFA (Graphical Fragment Assembly) and edges from Hi-C reads [35].
- 3D-DNA uses Hi-C reads to refine provided contigs before executing clustering, sorting, and orientation steps [35].
- ALLHiC is specifically designed for polyploid genomes, addressing the challenges of haplotype divergence [35].
- LACHESIS was the pioneering Hi-C scaffolding tool but requires pre-specification of chromosome numbers and is no longer under active development [34] [35].
Performance Evaluation Across Genome Types
Recent benchmarking studies provide comprehensive comparisons of Hi-C scaffolding tools. In haploid genome assembly, ALLHiC and YaHS achieved the highest completeness rates (99.26% and 98.26% respectively), significantly outperforming alternatives [35]. LACHESIS showed reasonable completeness (87.54%), while pin_hic, 3d-DNA, and SALSA2 had lower performance (55.49%, 55.83%, and 38.13% respectively) [35].
A 2024 study evaluating Hi-C tools for plant genomes found YaHS to be the best-performing tool, considering contiguity, completeness, accuracy, and structural correctness [34]. The performance of these tools is heavily influenced by the quality of the initial contig assembly, with studies highlighting that problems in initial assemblies cannot be resolved accurately by Hi-C data alone [1].
Figure 1: Hi-C Scaffolding Workflow. This diagram illustrates the key steps in Hi-C-based scaffolding, from initial contigs and Hi-C reads to final chromosome-scale assembly.
Table 3: Hi-C Scaffolding Tool Performance
| Scaffolder | Completeness (Haploid) | Correctness (PLC) | Key Features | Limitations |
|---|---|---|---|---|
| YaHS | 98.26% [35] | >99.8% [35] | Most accurate in recent benchmarks [34] | - |
| ALLHiC | 99.26% [35] | 98.14% [35] | Designed for polyploid genomes [35] | Lower correctness than YaHS |
| LACHESIS | 87.54% [35] | >99.8% [35] | Pioneer in Hi-C scaffolding [35] | Requires chromosome number; no longer developed [34] |
| 3D-DNA | 55.83% [35] | >99.8% [35] | Error correction before scaffolding [35] | Lower completeness |
| SALSA2 | 38.13% [35] | 94.96% [35] | Hybrid graph approach [35] | Lowest completeness |
Quality Assessment and Validation
Comprehensive quality assessment throughout the assembly workflow is crucial for generating high-quality genome assemblies. Standard metrics include:
- Contiguity metrics: N50/NG50 and L50/LG50 values measure assembly fragmentation [4].
- Completeness metrics: BUSCO (Benchmarking Universal Single-Copy Orthologs) assesses gene space completeness [4].
- Repeat space evaluation: LTR Assembly Index (LAI) estimates completeness of repetitive regions by assessing intact LTR retrotransposons [4].
- k-mer completeness: Merqury evaluates assembly accuracy using k-mer spectra [34].
Integrated tools like GenomeQC provide a comprehensive framework combining multiple quality metrics, enabling comparison against gold standard references and benchmarking across assemblies [4]. These assessments should be implemented throughout genome assembly pipelines, not just upon completion, to inform decisions and identify potential issues early [1].
Integrated Workflow Design
Based on the collective benchmarking evidence, an optimal integrated workflow would include:
- Data Acquisition: Combine ONT or PacBio long-read data (≥30× coverage) with Hi-C data and Illumina short reads for polishing.
- Assembly: Use Flye for an optimal balance of contiguity and computational efficiency, particularly with Ratatosk error-corrected long reads [33].
- Polishing: Apply two rounds of Racon followed by Pilon polishing for maximum accuracy [33].
- Scaffolding: Implement YaHS for Hi-C-based scaffolding, as it demonstrates superior performance in recent benchmarks [34] [35].
- Validation: Use multiple quality metrics (BUSCO, LAI, Merqury) throughout the process with tools like GenomeQC [4].
Figure 2: Optimal Integrated Workflow. This diagram outlines the recommended phases and tool selections for chromosome-scale genome assembly based on benchmarking studies.
The Scientist's Toolkit
Table 4: Essential Research Reagents and Computational Tools
| Resource | Category | Function | Example Tools/Datasets |
|---|---|---|---|
| Long-read Sequencer | Sequencing Platform | Generates long reads for assembly spanning repeats | Oxford Nanopore PromethION, PacBio Sequel [31] |
| Hi-C Library Kit | Library Preparation | Enables proximity ligation for chromatin interaction data | Dovetail Hi-C Kit, Arima Hi-C Kit |
| Assembly Software | Computational Tool | Constructs contiguous sequences from raw reads | Flye, Canu, WTDBG2 [1] [31] |
| Hi-C Scaffolder | Computational Tool | Orders and orients contigs into chromosomes | YaHS, SALSA2, 3D-DNA [34] [35] |
| Polishing Tools | Computational Tool | Corrects errors in draft assemblies | Racon, Medaka, Pilon [1] [33] |
| Quality Metrics | Assessment Tool | Evaluates assembly completeness and accuracy | BUSCO, Merqury, LAI [4] |
| Reference Genomes | Validation Resource | Benchmarking against known assemblies | NCBI Assembly Database [4] |
The integration of long-read sequencing technologies with Hi-C scaffolding has dramatically improved our ability to generate chromosome-scale genome assemblies. Benchmarking studies consistently show that tool selection significantly impacts final assembly quality, with Flye generally outperforming other assemblers, particularly when combined with Racon and Pilon polishing, and YaHS emerging as the superior Hi-C scaffolding tool in recent evaluations. Successful genome projects implement comprehensive quality assessment throughout the workflow rather than just upon completion, utilizing multiple complementary metrics to evaluate both gene space and repetitive regions. As sequencing technologies continue to evolve and computational methods advance, these integrated workflows will become increasingly accessible, enabling more researchers to generate high-quality genome assemblies for non-model organisms across diverse biological and biomedical research domains.
Genome assembly is a foundational step in genomics, profoundly influencing downstream applications in research, drug discovery, and personalized medicine. The choice of assembly pipeline—encompassing sequencing technologies, assemblers, and scaffolding methods—directly determines the contiguity, completeness, and accuracy of the resulting genomic sequence. This case study objectively benchmarks successful assembly pipelines across the plant, animal, and human domains, synthesizing current experimental data to provide a rigorous comparison for researchers and drug development professionals. Framed within the broader thesis of benchmarking genome assemblers, this guide summarizes performance characteristics, provides detailed experimental protocols, and visualizes key workflows to inform pipeline selection for diverse genomic projects.
Assembly Pipeline for Microbial Genomes
Microbial genomics requires efficient and accurate tools to reconstruct genomes for applications in pathogen surveillance, comparative genomics, and functional annotation.
Benchmarking of Long-Read Assemblers
A comprehensive benchmark of eleven long-read assemblers using standardized computational resources provides critical performance data [21]. The assemblers were evaluated on runtime, contiguity (N50, total length, contig count), GC content, and completeness using Benchmarking Universal Single-Copy Orthologs (BUSCO) [21].
Table 1: Performance Benchmark of Long-Read Microbial Genome Assemblers [21]
| Assembler | Runtime | Contiguity (N50) | BUSCO Completeness | Misassembly Rate | Key Characteristics |
|---|---|---|---|---|---|
| NextDenovo | Moderate | High | Near-complete | Low | Progressive error correction, consensus refinement; stable across preprocessing types |
| NECAT | Moderate | High | Near-complete | Low | Progressive error correction, consensus refinement; stable across preprocessing types |
| Flye | Moderate | High | High | Low | Balanced accuracy and contiguity; sensitive to corrected input |
| Canu | Very Long | Moderate (3-5 contigs) | High | Low | High accuracy but fragmented assemblies; longest runtimes |
| Unicycler | Moderate | Moderate | High | Low | Reliably produces circular assemblies; slightly shorter contigs |
| Raven | Fast | Moderate | Moderate | Moderate | — |
| Shasta | Ultrafast | Variable | Moderate (requires polishing) | Moderate | Highly dependent on preprocessing |
| Miniasm | Ultrafast | Variable | Moderate (requires polishing) | Moderate | Highly dependent on preprocessing |
| wtdbg2 (Redbean) | Fast | Low | Underperformed | Moderate | Structural instability and fragmentation |
| HINGE | Moderate | Low | Underperformed | Moderate | Underperformed |
Experimental Protocol for Microbial Assembly Benchmarking
The benchmarking study employed the following standardized methodology to ensure fair and reproducible comparisons [21]:
- Computational Resources: All assemblers were run using standardized computational resources to eliminate hardware performance bias.
- Evaluation Metrics: Assemblies were evaluated on a comprehensive set of metrics:
- Runtime: Total computational time required.
- Contiguity: Assessed via N50 statistic and total contig count.
- Completeness: Measured using Benchmarking Universal Single-Copy Orthologs (BUSCO) to identify conserved single-copy genes.
- Accuracy: Evaluated through GC content analysis and misassembly rates.
- Preprocessing Impact: The influence of different preprocessing methods (filtering, trimming, correction) on final assembly quality was systematically tested.
- Key Findings: Assemblers employing progressive error correction with consensus refinement (notably NextDenovo and NECAT) consistently generated near-complete, single-contig assemblies with low misassemblies. Preprocessing had a marked effect, with filtering improving genome fraction and BUSCO completeness, while correction sometimes increased misassemblies in graph-based tools [21].
Figure 1: Microbial Genome Assembly and Benchmarking Workflow. This diagram outlines the key steps for assembling and evaluating microbial genomes, from preprocessing of raw reads to assembly and final quality assessment.
Assembly Pipeline for Plant Genomes
Plant genomes present unique challenges, including large sizes, high ploidy, and abundant repetitive elements, necessitating specialized assembly and scaffolding strategies.
Chromosome-Level Assembly of Camellia rubituberculata
A chromosome-level genome assembly of Camellia rubituberculata, a species endemic to karst habitats, demonstrates a successful plant genomics pipeline. The assembly achieved a size of 2.50 Gb with 15 pseudo-chromosomes and a scaffold N50 of 168.34 Mb, annotating 55,302 protein-coding genes [36]. Comparative genomics revealed two whole-genome duplications, and selective sweep analysis identified genes associated with karst adaptation, including those involved in calcium homeostasis and ion transport [36].
Benchmarking Hi-C Scaffolding Tools for Plant Genomes
Hi-C scaffolding is crucial for achieving chromosome-level assemblies. A recent study benchmarked three Hi-C scaffolders—3D-DNA, SALSA2, and YaHS—using Arabidopsis thaliana assemblies from PacBio HiFi and Oxford Nanopore Technologies (ONT) data [37].
Table 2: Performance of Hi-C Scaffolding Tools on a Plant Genome [37]
| Scaffolder | Development Status | Accuracy in Ordering | Structural Correctness | Key Findings |
|---|---|---|---|---|
| YaHS | Most recently released | Highest | Highest | Best-performing tool in this benchmark |
| SALSA2 | Active development (successor to SALSA) | Moderate | Moderate | — |
| 3D-DNA | Widespread use, active development | Lower | Lower | — |
The experimental protocol for this benchmarking was as follows [37]:
- Data Source: Publicly available raw data from BioProject PRJCA005809, including PacBio HiFi, ONT, and Hi-C reads for Arabidopsis thaliana [37].
- Assembly Generation: Two distinct de novo assemblies were generated from the same raw data:
- Flye-based Assembly: ONT reads were assembled with Flye, polished with Racon using PacBio HiFi reads, and purged with Purgedups.
- Hifiasm-based Assembly: HiFi and ONT reads were assembled together using Hifiasm and purged with Purgedups.
- Scaffolding: Both assemblies were scaffolded using the three Hi-C scaffolders (3D-DNA, SALSA2, YaHS) with identical Hi-C data.
- Evaluation: The quality of the scaffolded assemblies was assessed using a custom Bash pipeline,
assemblyQC, which combined:- QUAST: For contiguity metrics without a reference.
- BUSCO: For completeness.
- Merqury: For quality evaluation.
- Liftoff: For annotation and analysis of gene positioning versus a reference genome.
Figure 2: Plant Genome Assembly and Hi-C Scaffolding Pipeline. The workflow for generating and benchmarking chromosome-level plant genome assemblies, highlighting the two primary assembly strategies and the evaluation of multiple Hi-C scaffolding tools.
Assembly Pipeline for Human Genomes
The completion of a telomere-to-telomere (T2T) human reference genome has set a new standard for accuracy and completeness, enabling more rigorous benchmarking of human genome assembly methods.
A Complete Diploid Human Genome Benchmark
A recent preprint presents a complete diploid genome benchmark for the HG002 individual, achieving near-perfect accuracy across 99.4% of the genome [38]. This benchmark adds 701.4 Mb of autosomal sequence and both sex chromosomes (216.8 Mb), totaling 15.3% of the genome absent from prior benchmarks [38]. It provides a diploid annotation of genes, transposable elements, segmental duplications, and satellite repeats, including 39,144 protein-coding genes across both haplotypes [38].
Performance of State-of-the-Art De Novo Assembly
This new benchmark was used to evaluate the performance of state-of-the-art sequencing and analysis methods. The analysis revealed that de novo assembly methods resolve 2-7% more sequence and outperform variant calling accuracy by an order of magnitude, yielding just one error per 100 kb across 99.9% of the benchmark regions [38]. This demonstrates the power of de novo assembly for generating complete and accurate personalized genomes, which is critical for advancing genomic medicine.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful genome assembly relies on a suite of specialized bioinformatics tools, sequencing technologies, and evaluation metrics.
Table 3: Essential Research Reagent Solutions for Genome Assembly
| Category | Item | Primary Function |
|---|---|---|
| Sequencing Technologies | PacBio HiFi Reads | Generates highly accurate long reads for resolving complex genomic regions [37]. |
| Oxford Nanopore Technologies (ONT) | Produces ultra-long reads for spanning repetitive elements and structural variants [37]. | |
| Illumina Short Reads | Provides high-accuracy short reads for polishing assemblies or variant calling [39]. | |
| Hi-C Sequencing | Enables chromosome-level scaffolding through proximity ligation data [37]. | |
| Assembly Software | Flye | Assembles long reads into contiguous sequences, effective with ONT data [37]. |
| Hifiasm | Efficiently assembles PacBio HiFi reads, often in combination with other data types [37]. | |
| NextDenovo | Produces near-complete, single-contig microbial assemblies via progressive error correction [21]. | |
| Scaffolding Tools | YaHS | Orders and orients contigs into scaffolds/chromosomes using Hi-C data; top performer in plant benchmarks [37]. |
| SALSA2 | Scaffolds genomes using Hi-C data; actively developed successor to SALSA [37]. | |
| 3D-DNA | A widely used Hi-C scaffolder; part of the popular Juicebox pipeline [37]. | |
| Quality Assessment | BUSCO | Assesses assembly completeness by benchmarking universal single-copy orthologs [21]. |
| QUAST | Evaluates assembly contiguity and quality with or without a reference genome [37]. | |
| Merqury | Measures assembly quality and phasing accuracy using k-mer spectra [37]. | |
| Data Sources | Biobanks (e.g., UK Biobank) | Provides large-scale, phenotypically rich genomic datasets for training AI models and discovery [40]. |
This case study demonstrates that while the core principles of genome assembly are universal, optimal pipeline design is highly specific to the biological domain. For microbial genomes, assemblers with progressive error correction like NextDenovo and NECAT provide the most complete and contiguous results. For complex plant genomes, combining long reads from PacBio HiFi or ONT with Hi-C scaffolding using YaHS is the most effective path to chromosome-scale assembly. For the ultimate in accuracy for human genomes, de novo assembly methods are now outperforming mapping-based approaches, as validated by the new complete diploid benchmark.
The field is moving toward more integrated, automated, and standardized pipelines, supported by benchmarks like those for Hi-C scaffolding and the complete human genome. The continued development of advanced benchmarking resources and tools will be crucial for empowering researchers and clinicians to generate the high-quality genomic data needed to unlock the full potential of personalized medicine and functional genomics.
Beyond the Basics: Optimizing Assembly Quality and Overcoming Common Challenges
In the realm of genomics, the adage "garbage in, garbage out" holds profound significance. The journey from raw sequencing data to a completed genome assembly is fraught with technical challenges, where the initial quality of the sequence reads critically influences all downstream analyses. Read filtering and trimming, collectively known as preprocessing, serve as the essential gatekeepers in this process, directly determining the accuracy, completeness, and contiguity of genome assemblies. Within the broader context of benchmarking genome assemblers, preprocessing emerges not as a mere preliminary step but as a decisive factor that can alter performance outcomes and subsequent biological interpretations. This guide objectively examines how preprocessing methodologies interact with various assembly tools, drawing on current experimental data to provide researchers, scientists, and drug development professionals with evidence-based recommendations for optimizing their genomic workflows.
The Fundamentals of Sequencing Data Preprocessing
Sequencing data preprocessing encompasses a series of computational operations designed to improve read quality before assembly. The process begins with quality assessment using tools like FastQC, which generates diagnostic plots visualizing per-base quality scores across all reads. These plots display quality distributions through box-and-whisker plots at each base position, with color-coded backgrounds (green, yellow, red) indicating quality ranges and helping researchers identify problematic regions [41].
The core preprocessing operations include:
- Filtering: The removal of entire reads that fail to meet quality thresholds, such as those containing an excess of ambiguous bases (N's) or exhibiting overall low quality scores [42].
- Trimming: The selective removal of adapter sequences, barcodes, and low-quality regions from read termini while preserving the remaining high-quality segments [43].
- Error Correction: Particularly crucial for long-read technologies like Oxford Nanopore, this process identifies and rectifies systematic basecalling errors through consensus methods [21].
Different sequencing technologies demand specialized preprocessing approaches. For Illumina short reads, tools like Trimmomatic implement algorithms such as SLIDINGWINDOW (which cuts reads when average quality within a window falls below a threshold) and HEADCROP (which removes a specified number of bases from read starts) [42]. For Nanopore long reads, SeqKit performs quality-based filtering, while NanoPlot provides quality assessment visualizations specific to long-read characteristics [41].
Table 1: Essential Preprocessing Tools and Their Functions
| Tool Name | Sequencing Technology | Primary Function | Key Parameters |
|---|---|---|---|
| FastQC | Illumina, Nanopore | Quality assessment and visualization | Per-base quality, adapter content, GC content |
| Trimmomatic | Illumina | Read trimming and filtering | SLIDINGWINDOW, HEADCROP, MINLEN |
| SeqKit | Nanopore | Quality-based read filtering | Quality threshold, read length |
| NanoPlot | Nanopore | Long-read quality assessment | Read length distribution, quality plots |
Experimental Protocols for Preprocessing and Assembly Benchmarking
Standardized experimental protocols are essential for rigorous benchmarking of how preprocessing influences genome assembly outcomes. The following methodology outlines a comprehensive approach derived from current literature:
Sample Preparation and Sequencing
High-molecular-weight DNA should be extracted using established protocols, such as the CTAB-based method for plant tissues or column-based systems for microbial cultures [44]. The extracted DNA must undergo quality control through spectrophotometric analysis and gel electrophoresis to ensure integrity and purity. Sequencing should be performed on both short-read (Illumina) and long-read (Oxford Nanopore or PacBio) platforms for the same biological sample to enable hybrid assembly comparisons [44].
Preprocessing Workflow
- Quality Assessment: Run FastQC on raw Illumina reads and NanoPlot on raw Nanopore reads to establish baseline quality metrics [41].
- Adapter Trimming: Remove sequencing adapters and barcodes using Trimmomatic for Illumina reads and Porechop for Nanopore reads.
- Quality Trimming: Apply Trimmomatic's SLIDINGWINDOW approach (e.g., 4-base window, 15 quality threshold) for Illumina reads [42]. For Nanopore reads, use quality filtering with SeqKit at a predetermined Q-score threshold [41].
- Length Filtering: Discard reads shorter than a minimum length (e.g., 100 bp for Illumina, 1 kb for Nanopore) using the MINLEN function in Trimmomatic or similar parameters in long-read tools [42].
- Error Correction: For long-read assemblers that benefit from pre-corrected input, apply error correction methods such as Canu's built-in correction or the Ratatosk tool [33].
Assembly and Evaluation
Assemble the preprocessed reads using multiple assemblers with standardized computational resources. Recommended assemblers include Flye, Raven, Canu, Miniasm/Racon, and Shasta for long-read data, with Unicycler for hybrid approaches [45] [21] [33]. Evaluate assemblies using QUAST for contiguity metrics (N50, contig count), BUSCO for completeness, and Merqury for accuracy assessment [33]. Additionally, validate assemblies through comparison with known reference genomes when available.
The following workflow diagram illustrates the complete experimental protocol from raw sequencing data to assembly evaluation:
Comparative Analysis of Preprocessing Impacts on Assembly Performance
Recent benchmarking studies reveal how preprocessing strategies significantly influence the performance of various genome assemblers. The interaction between read quality and assembly algorithm choice produces markedly different outcomes in terms of completeness, accuracy, and computational efficiency.
Preprocessing Effects on Long-Read Assemblers
A comprehensive benchmark of eleven long-read assemblers demonstrated that preprocessing steps—particularly filtering, trimming, and correction—jointly determine accuracy, contiguity, and computational efficiency [21]. Assemblers employing progressive error correction with consensus refinement (NextDenovo and NECAT) consistently generated near-complete, single-contig assemblies with low misassemblies across different preprocessing types. Flye offered a strong balance of accuracy and contiguity but was sensitive to corrected input, performing optimally with preprocessed reads [21]. Canu achieved high accuracy but produced fragmented assemblies (3–5 contigs) and required the longest runtimes, with preprocessing steps significantly impacting its resource consumption.
Ultrafast tools like Miniasm and Shasta provided rapid draft assemblies yet were highly dependent on preprocessing, requiring polishing to achieve completeness [21]. Specifically, filtered reads improved genome fraction and BUSCO completeness, while trimming reduced low-quality artifacts. Correction benefited overlap-layout-consensus (OLC)-based assemblers but occasionally increased misassemblies in graph-based tools [21].
Table 2: Assembly Performance with Different Preprocessing Approaches
| Assembler | Algorithm Type | Optimal Preprocessing | Contiguity (N50) | Completeness (BUSCO) | Runtime |
|---|---|---|---|---|---|
| NextDenovo | OLC with refinement | Filtering + Correction | High | Near-complete | Moderate |
| Flye | Repeat graph | Quality trimming | High | Complete | Moderate |
| Raven | OLC | Minimal preprocessing | Medium-high | Complete | Fast |
| Canu | OLC | Built-in correction | Medium | Complete | Very slow |
| Miniasm/Racon | OLC | Correction + Polishing | Medium | High (with polishing) | Very fast |
| Shasta | Run-length | Quality filtering | Medium | Medium (requires polishing) | Fastest |
Impact on Downstream Genomic Analyses
The influence of preprocessing extends beyond assembly metrics to critical downstream applications. In benchmarking studies focused on bacterial pathogens, preprocessing quality directly affected the accuracy of antimicrobial resistance (AMR) profiles, virulence gene prediction, and multilocus sequence typing (MLST) [45]. All Miniasm/Racon and Raven assemblies of mediocre-quality reads provided accurate AMR profiles, while only the Raven assembly of Klebsiella variicola with low-quality reads yielded an accurate AMR profile across all assemblers and species [45]. Regarding virulence genes, all assemblers functioned well with mediocre-quality and real reads, whereas only Raven assemblies of low-quality reads maintained accurate numbers of virulence genes after preprocessing.
For phylogenetic inference and pan-genome analyses, Miniasm/Racon and Raven assemblies demonstrated the most accurate performance, highlighting how appropriate preprocessing enables reliable biological interpretations [45]. These findings underscore that preprocessing choices should be guided by the specific downstream applications planned for the assembled genomes.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful genome assembly projects require both computational tools and wet-laboratory reagents that work in concert to produce high-quality data. The following table details essential solutions for sequencing and assembly workflows:
Table 3: Essential Research Reagents and Computational Tools
| Item | Function | Application Notes |
|---|---|---|
| High-Molecular-Weight DNA Extraction Kit | Isolate intact genomic DNA | Critical for long-read sequencing; maintain DNA integrity |
| PCR Barcoding Expansion Kit | Multiplex samples | Allows sequencing of multiple samples in a single run |
| SMRTbell Template Prep Kit | Library preparation for PacBio | Optimized for long-insert libraries |
| Ligation Sequencing Kit | Library preparation for Nanopore | Enables direct RNA or DNA sequencing |
| AMPure PB Beads | DNA size selection and purification | Remove short fragments and purify reactions |
| Trimmomatic | Read trimming and filtering | Flexible parameters for Illumina data [42] |
| SeqKit | Nanopore read processing | Fast quality-based filtering of long reads [41] |
| FastQC | Quality control visualization | First step in any preprocessing pipeline [41] |
| Racon | Consensus polishing | Improves assembly accuracy after initial assembly [33] |
| Pilon | Polish assemblies with short reads | Uses Illumina data to correct systematic errors |
Preprocessing of sequencing reads through filtering and trimming represents a critical determinant in the success of genome assembly projects. The experimental data presented in this comparison guide consistently demonstrates that preprocessing choices directly influence assembly contiguity, completeness, and accuracy across diverse benchmarking studies. The interaction between preprocessing methods and assembler algorithms is complex, with no single universally optimal approach, yet clear patterns emerge. Methods incorporating progressive error correction with consensus refinement generally produce superior results, particularly when combined with quality-aware trimming and filtering. For researchers and drug development professionals, establishing standardized preprocessing protocols tailored to their specific biological questions and sequencing technologies will yield more reliable genomic assemblies and subsequent analyses. As sequencing technologies continue to evolve, ongoing benchmarking of preprocessing and assembly pipelines remains essential for advancing genomic research and its clinical applications.
In the field of genomic research, the reconstruction of complete and accurate genomes from sequencing data remains a foundational challenge. Long-read sequencing technologies, particularly those from Oxford Nanopore Technologies (ONT), have revolutionized de novo assembly by producing reads that can span complex repetitive regions, leading to highly contiguous genomes [14] [46]. However, these reads often possess a high inherent error rate, typically between 5% and 15%, which can result in several thousand base errors in a typical bacterial genome assembly [14] [47]. Genome polishing addresses this critical limitation by employing computational tools to correct nucleotide errors in draft assemblies, a step that is indispensable for applications requiring ultra-high accuracy, such as outbreak source tracking, genetic variant discovery, and gene annotation [14] [48].
This guide is situated within a broader thesis on benchmarking genome assembly and refinement workflows. The performance of polishing tools is not absolute but is significantly influenced by factors such as the choice of assembler, sequencing coverage, genomic context (e.g., homopolymer tracts), and, most importantly, the specific combination and order of tools used in a pipeline [14] [21]. A benchmarking study on Salmonella enterica outbreak isolates underscored that while long-read polishing alone enhances accuracy, achieving "near-perfect" genomes (exceeding 99.9999% accuracy) often necessitates a hybrid approach that leverages both long- and short-read data [14] [49]. This article provides a structured comparison of three cornerstone tools in the polishing landscape—Racon, Medaka, and Pilon—by synthesizing recent experimental data to offer evidence-based guidance for researchers and drug development professionals.
Racon is a long-read polisher that performs rapid consensus calling based on overlapped sequences. It is designed to be used as a standalone polisher and can be iteratively applied. However, its performance is often superseded by more recent tools [14] [47].
Medaka is a long-read polishing tool developed by Oxford Nanopore Technologies that employs a neural network model trained on specific sequencing chemistry error profiles. It is noted for being more accurate and computationally efficient than Racon [14]. It is important to note that ONT has since released a successor to Medaka, the dorado polish tool, which is designed to work seamlessly with the latest basecalling models [50].
Pilon is a widely used short-read polisher that utilizes high-accuracy Illumina data to correct base errors, fill gaps, and fix misassemblies in a draft assembly. It is particularly effective for correcting single-nucleotide errors (SNPs) but can introduce errors in repetitive regions where short reads cannot be uniquely mapped [14] [33].
Table 1: Summary of Polishing Tool Performance Based on Benchmarking Studies
| Tool | Read Type | Key Strengths | Key Limitations | Reported Performance (vs. Reference Genome) |
|---|---|---|---|---|
| Racon | Long-read | Fast consensus calling; can be used iteratively. | Less accurate than Medaka; performance is pipeline-dependent. | Higher error rates compared to Medaka-polished assemblies [14]. |
| Medaka | Long-read | High accuracy and efficiency; uses ONT-specific error models. | Requires specific model for sequencing chemistry; being superseded by dorado polish [50]. |
More accurate and efficient than Racon; reduced errors in draft assemblies [14] [50]. |
| Pilon | Short-read | Highly effective at correcting SNPs and small indels. | Can introduce errors in repetitive/low-complexity regions. | Similar accuracy to NextPolish and POLCA; performance relies on long-read polishing first [14]. |
Comparative Experimental Data and Workflow Analysis
Key Findings from Benchmarking Studies
A comprehensive benchmark evaluating 132 polishing pipelines for Salmonella Newport genomes revealed critical insights into the performance of Racon, Medaka, and Pilon [14] [49]. The study established that while long-read polishing alone improves assembly quality, the highest accuracy—approaching 99.9999% or about 5 errors in a 4.8 Mbp genome—was only attained through combined long- and short-read polishing [14]. In direct comparisons, Medaka proved to be a more accurate and efficient long-read polisher than Racon [14]. Among short-read polishers, Pilon demonstrated high accuracy, performing similarly to other tools like NextPolish and Polypolish [14].
The order of tool application was found to be critical. The benchmark showed that applying a less accurate tool after a more accurate one can reintroduce errors. Consequently, the most successful pipelines consistently used Medaka for long-read polishing prior to Pilon for short-read polishing, rather than the reverse order [14]. A separate, independent benchmarking study on human genome assembly confirmed this finding, reporting that two rounds of Racon followed by Pilon yielded the best results for that specific dataset and tool combination [33] [5]. This underscores that the optimal pipeline can vary based on the organism and data type.
Table 2: Summary of Optimal Polishing Strategies from Recent Studies
| Organism/Context | Recommended Polishing Workflow | Reported Outcome | Citation |
|---|---|---|---|
| Salmonella Newport (Bacterial Outbreak) | Flye assembly → Medaka → NextPolish (or Pilon) | Achieved near-perfect accuracy (~5 errors/genome); order was critical. | [14] |
| Human Genome (HG002) | Flye assembly → 2x Racon → Pilon | Yielded the best assembly accuracy and continuity. | [33] [5] |
| Pseudomonas aeruginosa (Clinical Isolate) | Flye assembly → dorado correct (pre-assembly) → dorado polish (post-assembly) | Achieved high concordance with Illumina references (as few as 2 discordant positions). | [50] |
Experimental Protocols from Cited Studies
Protocol 1: Hybrid Polishing for Bacterial Outbreak Isolates [14]
- Assembly: Generate an initial draft assembly from nanopore reads using a long-read assembler like Flye.
- Long-read Polishing: Polish the draft assembly with Medaka. This involves mapping the nanopore reads back to the assembly using Minimap2 and then running Medaka with the appropriate model that matches your sequencing chemistry (e.g.,
r941_prom_sup_g507for high-accuracy R9.4.1 pores). - Short-read Polishing: Further polish the Medaka-corrected assembly with Pilon. This requires mapping high-quality Illumina reads to the polished assembly using BWA-MEM or Bowtie2. Pilon is then run on the resulting alignment file to make base-level corrections.
- Validation: Evaluate the final polished assembly by comparing it to a high-quality reference genome (e.g., a PacBio HiFi assembly) to quantify remaining errors.
Protocol 2: Iterative Racon and Pilon for Human Genome [33] [5]
- Assembly and Initial Correction: Assemble ONT reads with Flye. Optionally, perform pre-assembly error correction of the reads with a tool like Ratatosk.
- Iterative Long-read Polishing: Run Racon for two iterative cycles. Each cycle involves mapping the reads to the current assembly with Minimap2 and then running Racon with parameters
-m 8 -x -6 -g -8 -w 500to generate a new consensus. - Short-read Polishing: Perform final polishing with Pilon using high-coverage Illumina reads to correct remaining small errors.
Visualization of Optimal Polishing Workflows
The following workflow diagram synthesizes the most effective polishing strategies identified in the benchmarking studies.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Genome Polishing Experiments
| Item Name | Function/Application | Specific Example/Note |
|---|---|---|
| ONT Sequencing Kit | Generates long-read data for assembly and long-read polishing. | Ligation Sequencing Kit (SQK-LSK109) [47] [46]. |
| ONT Flow Cell | The consumable device through which DNA is sequenced. | Choose chemistry appropriate for accuracy needs (e.g., R9.4.1, R10.4.1) [50]. |
| Illumina Sequencing Kit | Generates high-accuracy short-read data for hybrid polishing. | Nextera DNA Flex Library Prep or MiSeq Reagent Kit [14] [47]. |
| High-Molecular-Weight (HMW) DNA Extraction Kit | To obtain long, intact DNA fragments optimal for long-read sequencing. | Critical for achieving long read lengths and high N50 [46]. |
| Reference Genome Material | Provides a gold standard for benchmarking and validating polishing accuracy. | e.g., Genome in a Bottle (GIAB) human reference [5] or organism-specific PacBio HiFi assembly [14]. |
The evidence from recent benchmarking studies leads to several clear recommendations for researchers aiming to maximize base-level accuracy. First, a hybrid approach combining long-read and short-read polishing is the most reliable path to near-perfect genomes. Relying solely on long-read data, even with advanced tools like Medaka, may not suffice for applications like outbreak tracking where single-nucleotide differences are meaningful [14] [49].
Second, tool order is not a trivial detail. The consensus from the literature strongly advises performing long-read polishing with Medaka before applying short-read correction with Pilon [14]. While an iterative Racon and Pilon approach has proven effective for human genomes [33] [5], Medaka generally outperforms Racon in bacterial genomics contexts and is more efficient [14]. Finally, researchers must stay abreast of tool development. Newer integrated pipelines like the dorado suite (which includes dorado correct and dorado polish) are emerging as promising successors, potentially simplifying workflows while maintaining or improving accuracy [50]. By strategically combining these tools and following validated protocols, scientists can build a robust foundation for all downstream genomic analyses.
Eukaryotic genome assembly presents formidable challenges, primarily due to pervasive repetitive elements and diploid nature. Repetitive DNA sequences constitute 25-50% of mammalian genomes, while heterozygous regions complicate the resolution of individual haplotypes [51]. These complex architectures create algorithmic bottlenecks for assemblers, particularly in resolving long tandem repeats, segmental duplications, and transposable elements [52] [51]. The limitations are most pronounced in clinical contexts, where incomplete resolution of repetitive regions can obscure structural variants implicated in diseases [53] [54].
Recent technological advances in long-read sequencing and specialized assembly algorithms have begun to address these challenges. This comparison guide objectively evaluates the performance of contemporary genome assemblers and sequencing platforms in resolving haplotypes and repetitive regions, providing researchers with evidence-based selection criteria for their specific genomic applications.
Comparative Performance Analysis of Sequencing and Assembly Strategies
Sequencing Platform Performance Characteristics
Table 1: Comparison of Sequencing Technologies for Complex Genome Assembly
| Sequencing Technology | Read Length (N50) | Raw Accuracy | Systematic Biases | Best Application Context |
|---|---|---|---|---|
| PacBio HiFi | 13-20 kb | >99.9% (QV40) [53] | Low GC bias [31] | Haplotype-resolved assemblies, variant detection [53] [55] |
| Oxford Nanopore | 20-77 kb [53] | ~97% (QV30) [31] | Homopolymer indels [31] | Structural variant detection, long repeat resolution [53] |
| Illumina NovaSeq 6000 | 150-300 bp | >99.5% (QV40) [31] | GC bias, limited in repeats [31] | Polishing, validation, expression analysis [53] |
| DNBSEQ-T7 | 100-300 bp | High (comparable to Illumina) [31] | Similar to Illumina [31] | Cost-effective polishing [31] |
Assembler Performance Across Genomic Contexts
Table 2: Performance Comparison of Genome Assemblers on Challenging Regions
| Assembly Tool | Algorithm Type | Contiguity (Contig N50) | Repeat Resolution Capability | Haplotype Resolution | Computational Demand |
|---|---|---|---|---|---|
| Hifiasm | De novo/Optimized for HiFi | 26-133 Mb [53] [54] | Excellent for segmental duplications [53] | Yes (haplotype-resolved) [53] [56] | Moderate [56] |
| Flye | De novo/Optimized for long reads | High (comparable to Hifiasm) [53] | Graph-based repeat resolution [31] | Limited without additional data [53] | Fast [31] |
| Canu | De novo/Optimized for noisy reads | High | Repeat-sensitive overlap [31] | Limited without additional data | High (multiple corrections) [31] |
| Shasta | De novo/Optimized for Nanopore | High | Moderate | No | Fast [53] |
| MaSuRCA | Hybrid | Moderate | Good with hybrid approach | Limited | Moderate [31] |
Performance on Specific Genomic Challenges
Table 3: Assembly Performance Across Specific Genomic Challenges
| Genomic Challenge | Best Performing Approach | Key Metrics | Limitations |
|---|---|---|---|
| Centromeric satellites | ONT ultra-long reads (77 kb N50) [53] | Closed 236-251 GRCh38 gaps [53] | Requires high DNA quantity and quality |
| Segmental duplications | PacBio HiFi with Hifiasm [54] | 95.43% coverage of accessible regions [54] | Higher cost per sample |
| Large inversions (>4 Mb) | Combined long-read assembly [53] | Resolved polymorphic inversions [53] | Requires complementary data for phasing |
| Mobile element insertions | Phased assembly variant calling [54] | Identified 68% SVs missed by short-reads [54] | Computational complexity |
| Highly heterozygous regions | Hifiasm with Hi-C integration [56] | Achieved 377 Mb/343 Mb haplotypes [56] | Requires additional library preparation |
Experimental Protocols for Assembly Benchmarking
Comprehensive Benchmarking Workflow for Assemblers
Chinese Quartet Benchmarking Study Protocol
The Chinese Quartet study established a robust benchmarking protocol for assessing assembler performance on complex genomic regions [53]:
Sample Design:
- Two monozygotic twin daughters and their biological parents
- Certified Reference Materials approved by State Administration for Market Regulation in China
- Enables distinction between genuine variants and technical artifacts
Sequencing Approach:
- Approximately 50× PacBio HiFi coverage (read N50: 13-14 kb)
- Approximately 100× Oxford Nanopore regular coverage (read N50: 20-25 kb)
- Approximately 30× Oxford Nanopore ultra-long coverage (read N50: 77 kb) for one twin
- Approximately 160× Illumina NovaSeq and 100× BGI short-read coverage
Assembly Strategy:
- Reads from monozygotic twins merged to improve haplotype resolution
- HiFi, regular ONT, and ultra-long ONT reads phased into paternal and maternal haplotypes
- Five different assembly approaches tested (Shasta, Flye, Hifiasm, Hicanu, Flye with HiFi)
- Final assembly polished with phased HiFi reads
Variant Cataloging:
- Comprehensive variant calling including 3,962,453 SNVs, 886,648 indels
- 9,726 large deletions (≥50 bp), 15,600 large insertions (≥50 bp)
- 40 inversions, 31 complex structural variants, 68 shared de novo mutations
Haplotype-Resolved Assembly Protocol for Diverse Human Genomes
The Human Genome Structural Variation Consortium established this protocol for generating haplotype-resolved assemblies without parent-child trios [54]:
Sample Diversity:
- 35 genomes representing 26 1000 Genomes Project populations
- 19 females and 16 males of African, Admixed American, East Asian, European, and South Asian descent
Sequencing and Phasing:
- Continuous Long Read (CLR) sequencing (>40× coverage) or HiFi sequencing (>20× coverage)
- Strand-seq data (74-183 cells per sample) for phasing
- Reference-free assembly approach
Quality Validation:
- Base-pair accuracy quantification (QV > 40)
- Contiguity assessment (contig N50 > 25 Mbp)
- Switch error rate calculation (median 0.12%)
- Optical mapping concordance verification (>97%)
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Solutions for Genome Assembly Studies
| Reagent/Resource | Specific Example | Function in Workflow | Technical Considerations |
|---|---|---|---|
| Long-read Sequencing Kit | SMRTbell prep kit 3.0 [55] | Library preparation for PacBio systems | Enables multiplexing up to 96 samples |
| DNA Extraction Kit | Nanobind HT CBB kit [55] | High-quality DNA extraction | Preserves long DNA fragments |
| Barcoded Adapters | SMRTbell barcoded adapter plate 3.0 [55] | Sample multiplexing | Reduces per-sample sequencing costs |
| Hi-C Library Kit | DpnII-based digestion [56] | Chromatin conformation capture | Enables chromosome-scale scaffolding |
| DNA Shearing System | Plate-based high-throughput shearing [55] | DNA fragmentation | 3-minute processing, <$1.00/sample |
| Reference Materials | Chinese Quartet DNA [53] | Method benchmarking | Certified reference materials |
| Validation Technologies | Bionano Genomics optical mapping [54] | Assembly validation | Provides orthogonal confirmation |
Discussion and Future Perspectives
The benchmarking data reveals that no single assembler consistently outperforms all others across every metric, emphasizing the importance of application-specific selection. For comprehensive variant discovery including complex structural variants, PacBio HiFi-based assemblies provide superior base-level accuracy and haplotype resolution [53] [54]. For applications prioritizing extreme contiguity in repeat-rich regions, Oxford Nanopore ultra-long reads offer advantages despite higher error rates [53] [31].
The emerging paradigm for excellence in genome assembly involves integrated approaches that combine multiple technologies. The most successful strategies use long-read technologies for initial assembly followed by short-read polishing for base-level accuracy, supplemented with Hi-C or Strand-seq for phasing and scaffolding [53] [54] [56]. This hybrid approach effectively addresses the dual challenges of repetitive regions and haplotype resolution.
Future developments will likely focus on algorithmic improvements for complex variant detection and cost reduction through streamlined protocols. The recently developed high-throughput microbial workflow demonstrates promising directions, achieving 4-12-fold throughput enhancements with per-sample costs below $1.00 [55]. Such advances will make comprehensive genome assembly more accessible across diverse research contexts.
For clinical applications, particularly in drug development and complex disease research, haplotype-resolved assemblies provide critical insights by enabling precise mapping of structural variants and their phase relationships [53] [54]. This capability is essential for understanding compound heterozygosity and cis-regulatory interactions that underlie disease mechanisms and therapeutic responses.
In the field of genomics, the reconstruction of complete genome sequences from raw sequencing data remains a foundational yet computationally intensive challenge. The choice of assembly tools and parameters directly influences downstream biological interpretations, impacting applications ranging from comparative genomics to drug target discovery [21]. With the continuous evolution of sequencing technologies and algorithmic methods, researchers face a complex landscape of assemblers, each with distinct performance characteristics regarding runtime, memory consumption, and output accuracy. This guide provides an objective comparison of modern genome assembly tools, framed within the broader context of computational resource management, to inform selection strategies for scientific researchers and drug development professionals.
The benchmarking philosophy in computational genomics recognizes that no single assembler is universally optimal; rather, the choice represents a series of trade-offs that must be balanced against project-specific constraints and objectives [21]. Comprehensive evaluations must consider multiple performance dimensions simultaneously, including computational efficiency measured through runtime and memory usage, and output quality assessed through contiguity, completeness, and error rates. This guide synthesizes findings from recent, rigorous benchmarking studies to provide evidence-based recommendations for tool selection across various genomic contexts.
Performance Metrics and Benchmarking Methodology
Standardized Metrics for Assembly Evaluation
The assessment of genome assemblers relies on standardized quantitative metrics that capture different aspects of performance. Contiguity metrics include the N50 statistic (the length of the shortest contig at 50% of the total assembly length) and total assembly size, which indicate how completely the assembly reconstructs the genome in large, contiguous pieces [21]. Completeness is typically evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO), which measures the presence of evolutionarily conserved genes, while the LTR Assembly Index (LAI) assesses the completeness of repetitive regions [4]. Accuracy metrics evaluate the rate of misassemblies and base-level errors, often quantified through quality value (QV) scores and k-mer completeness analysis [5] [4]. Computational efficiency is measured through runtime (often wall-clock time) and peak memory usage, which determine the practical feasibility of assembly projects [57].
Experimental Design in Benchmarking Studies
Robust benchmarking requires standardized experimental protocols to ensure fair comparisons. The Genome in a Bottle (GIAB) consortium provides reference materials and benchmark calls that enable consistent evaluation of assembly pipelines, particularly for human genomes [5] [58]. Best practices in benchmarking involve running assemblers on the same dataset using equivalent computational resources (CPU cores, memory) and evaluating outputs against a common truth set [21] [5]. Studies typically employ multiple datasets representing different sequencing technologies (ONT, PacBio, Illumina), coverage depths, and genome types to assess performance across diverse conditions. Evaluation workflows like those implemented in QUAST, BUSCO, and Merqury provide standardized assessment of the resulting assemblies [5] [4].
Table 1: Key Performance Metrics for Genome Assembly Evaluation
| Metric Category | Specific Metrics | Interpretation | Measurement Tools |
|---|---|---|---|
| Contiguity | N50, L50, NG50, Total length | Higher N50 indicates more contiguous assemblies | QUAST, GenomeQC |
| Completeness | BUSCO score (% complete), LAI | Higher scores indicate more complete gene space and repetitive regions | BUSCO, LTR_retriever |
| Accuracy | QV score, k-mer completeness, misassembly rate | Higher QV and k-mer completeness with lower misassembly rates indicate higher accuracy | Merqury, QUAST |
| Computational Efficiency | Wall-clock time, CPU time, Peak memory | Lower values indicate more efficient resource usage | Native system monitoring |
Comparative Performance of Genome Assemblers
Long-Read Assembler Benchmarking
Comprehensive evaluations of long-read assemblers reveal distinct performance profiles across different tools. A benchmark of eleven long-read assemblers using standardized computational resources found that assemblers employing progressive error correction with consensus refinement, notably NextDenovo and NECAT, consistently generated near-complete, single-contig assemblies with low misassemblies and stable performance across different preprocessing approaches [21]. Flye offered a strong balance of accuracy and contiguity, although it demonstrated sensitivity to corrected input data. Canu achieved high accuracy but produced more fragmented assemblies (3–5 contigs) and required the longest runtimes, making it computationally intensive [21]. Ultrafast tools such as Miniasm and Shasta provided rapid draft assemblies but were highly dependent on preprocessing and required additional polishing steps to achieve completeness.
The impact of preprocessing strategies significantly influences assembler performance. Filtering reads typically improves genome fraction and BUSCO completeness, while trimming reduces low-quality artifacts. Error correction generally benefits overlap-layout-consensus (OLC)-based assemblers but can occasionally increase misassemblies in graph-based tools [21]. These findings underscore that assembler choice and preprocessing strategies jointly determine the accuracy, contiguity, and computational efficiency of the final assembly.
Table 2: Performance Comparison of Long-Read Genome Assemblers
| Assembler | Runtime | Memory Usage | Contiguity (N50) | Completeness (BUSCO) | Best Use Case |
|---|---|---|---|---|---|
| NextDenovo | Medium | Medium | High | High | High-quality reference genomes |
| NECAT | Medium | Medium | High | High | High-quality reference genomes |
| Flye | Medium | Medium | High | High | Balanced projects |
| Canu | Very High | High | Medium | High | Accuracy-critical applications |
| Miniasm | Very Low | Low | Variable | Low without polishing | Rapid draft assemblies |
| Shasta | Very Low | Low | Variable | Low without polishing | Rapid draft assemblies |
| Unicycler | Medium | Medium | Medium | High | Circular assembly (plasmids, bacteria) |
Hybrid and Short-Read Assemblers
For hybrid approaches that combine long and short reads, benchmarking studies have demonstrated that polishing strategies significantly improve assembly accuracy and continuity. The optimal performing pipeline identified in recent research used Flye for initial assembly followed by two rounds of Racon and Pilon polishing, yielding the best results for human genome assembly [5]. This combination achieved superior metrics in QUAST, BUSCO, and Merqury evaluations, balancing computational demands with output quality.
In the metagenomics domain, MEGAHIT represents a highly optimized solution for large and complex metagenomic datasets. Utilizing a succinct de Bruijn graph data structure, MEGAHIT achieves memory-efficient assembly without compromising excessively on quality [59] [57]. Benchmarking experiments on the Iowa Prairie Soil dataset (252 Gbp) showed that MEGAHIT completed assembly in 43.5 hours using 243 GB memory, representing a favorable balance of computational requirements and assembly quality for metagenomic projects [59]. SPAdes remains another robust option, particularly for single-cell and standard multicell bacterial datasets, employing innovative algorithmic solutions to address challenges such as non-uniform coverage and chimeric reads [60].
Resource Management Strategies and Trade-offs
Computational Resource Requirements
Memory usage represents one of the most significant constraints in genome assembly, particularly for large eukaryotic genomes and complex metagenomes. Traditional de Bruijn graph-based assemblers require substantial RAM to construct and traverse the graph structure, with memory consumption scaling with genome size, complexity, and sequencing depth [59] [57]. Evaluations of metagenome assemblers on terabyte-sized datasets reveal distinct memory usage patterns, with MetaSPAdes consuming approximately 250 GB for a 233 GB wastewater metagenome dataset, while MEGAHIT demonstrated more efficient memory utilization through its succinct data structures [57].
Runtime considerations must account for both the computational complexity of assembly algorithms and their practical implementation. Tools like Miniasm and Shasta prioritize speed through simplified assembly approaches but sacrifice accuracy, typically requiring additional polishing steps [21]. In contrast, Canu's comprehensive error correction and consensus steps result in significantly longer runtimes but generally higher accuracy [21]. The relationship between runtime and accuracy is not always linear, with tools like Flye and NextDenovo offering favorable intermediate positions in this trade-off space.
Innovative Approaches to Resource Management
Persistent Memory (PMem) technology presents a promising approach to address memory limitations in large-scale assembly projects. PMem can effectively expand memory capacity beyond traditional DRAM constraints, enabling assembly of larger datasets than previously possible on a single node [57]. Performance evaluations demonstrate that PMem can substitute for DRAM with a variable impact on runtime; substituting up to 30% of total memory with PMem showed no appreciable slowdown, while 100% substitution resulted in approximately a 2.17× increase in runtime for metaSPAdes [57]. This trade-off between memory cost and computational speed provides a valuable strategy for resource-constrained environments.
For projects involving structural variant detection, the choice between alignment-based and assembly-based methods involves significant resource considerations. Assembly-based tools excel in detecting large insertions and demonstrate robustness to coverage fluctuations but demand substantially more computational resources [61]. Alignment-based methods offer superior computational efficiency and perform better at low sequencing coverage (5-10×) but may miss some complex variants [61]. This fundamental trade-off between detection power and resource requirements must be balanced according to the specific variant discovery goals of each project.
Experimental Protocols and Research Toolkit
Standardized Benchmarking Workflow
The following diagram illustrates a comprehensive benchmarking workflow for genome assemblers, integrating best practices from recent evaluations:
Diagram 1: Genome Assembler Benchmarking Workflow
Essential Research Reagent Solutions
Table 3: Key Bioinformatics Tools for Assembly Evaluation and Quality Control
| Tool Name | Function | Application Context |
|---|---|---|
| QUAST | Quality assessment tool for genome assemblies | Evaluates contiguity metrics (N50, L50) and identifies misassemblies |
| BUSCO | Benchmarking Universal Single-Copy Orthologs | Assesses completeness of gene space using evolutionarily conserved genes |
| Merqury | k-mer-based quality evaluation | Provides QV scores and k-mer completeness metrics for accuracy assessment |
| GenomeQC | Comprehensive quality assessment | Integrates multiple metrics including BUSCO, N50, and contamination checks |
| LTR_retriever | LTR Assembly Index (LAI) calculation | Evaluates completeness of repetitive regions in genome assemblies |
| Truvari | Structural variant comparison | Benchmarks SV calls against truth sets for validation |
The benchmarking data presented in this guide reveals that computational resource management in genome assembly requires careful consideration of the trade-offs between runtime, memory usage, and accuracy. For projects prioritizing assembly quality and completeness, particularly for reference-grade genomes, NextDenovo, NECAT, and Flye represent strong choices, with Flye offering a particularly balanced profile [21]. When computational resources are constrained, either in terms of time or memory, MEGAHIT provides an efficient solution for large datasets, while tools like Shasta and Miniasiasm offer ultra-fast draft assembly with the understanding that additional polishing will be required [21] [59].
The context of the assembly project significantly influences optimal tool selection. For bacterial genomes and small eukaryotes, where computational constraints are less pressing, Canu and Flye produce excellent results. For large, complex eukaryotic genomes, NextDenovo and NECAT offer robust performance, while for metagenomic datasets with high diversity and uneven coverage, MEGAHIT and metaSPAdes provide the necessary scalability [21] [59]. Emerging technologies like Persistent Memory (PMem) offer promising pathways to expand computational capabilities, potentially enabling more researchers to tackle larger and more complex assembly projects without prohibitive infrastructure investments [57].
As sequencing technologies continue to evolve and algorithmic improvements emerge, the landscape of genome assembly tools will undoubtedly change. The benchmarking framework and comparative approach outlined in this guide provide a foundation for researchers to evaluate new tools in the context of their specific project requirements, computational resources, and accuracy thresholds. By applying these evidence-based selection criteria, researchers and drug development professionals can optimize their computational resource allocation while maximizing the biological insights gained from their genome assembly projects.
Measuring Success: A Framework for Validation and Comparative Analysis of Assemblies
Reference-Free and Reference-Based Quality Assessment with QUAST and Merqury
Within the broader context of benchmarking genome assemblers, the selection of an appropriate quality assessment tool is as critical as the choice of the assembler itself. The rapid evolution of sequencing technologies, particularly long-read platforms, has produced assemblies that often surpass the quality of available reference genomes, rendering traditional validation methods insufficient [62]. This creates an urgent need for robust assessment methods that can objectively evaluate assembly quality without introducing reference bias. Two principal paradigms have emerged: reference-based methods, which compare assemblies to a known reference, and reference-free methods, which leverage intrinsic features of the data for evaluation.
QUAST (Quality Assessment Tool for Genome Assemblies) represents the reference-based approach, providing comprehensive metrics by aligning contigs to a reference genome. In contrast, Merqury adopts a reference-free methodology, utilizing k-mer-based analysis of unassembled high-accuracy reads to evaluate assembly quality and completeness [62]. This guide provides an objective comparison of these tools, detailing their operational principles, performance characteristics, and optimal use cases, supported by experimental data from recent benchmarking studies.
Tool Fundamentals: Operational Principles and Methodologies
QUAST: Reference-Based Assessment
QUAST functions by aligning assembled contigs to a pre-existing high-quality reference genome. This alignment forms the basis for calculating a suite of metrics that describe contiguity, completeness, and correctness. Key metrics include NA50 and NGA50 (which adjust N50 by considering alignments to the reference), misassembly counts, and the total aligned length. The fundamental strength of this approach is its ability to provide a direct, structural comparison to a ground truth. However, its major limitation is its dependence on a high-quality reference that may not be available for non-model organisms, and its potential to misclassify true biological variants in the assembled genome as errors [62].
Merqury: Reference-Free Assessment
Merqury circumvents the need for a reference genome by leveraging k-mers—unique substrings of length k—derived from high-accuracy sequencing reads (typically Illumina). Its core operation involves comparing the k-mers present in the assembly to those found in the unassembled read set [62] [63]. This comparison yields several critical metrics:
- Quality Value (QV): A Phred-scaled estimate of the consensus accuracy of the assembly, calculated from the proportion of k-mers found in the read set that are also present in the assembly.
- Completeness: The proportion of k-mers from the read set that are found in the assembly, indicating how much of the original sequence data is captured.
- k-mer Spectrum Plots: Visual representations that relate k-mer multiplicity in the read set to their copy number in the assembly, enabling the identification of issues like missing sequences or artificial duplications [62].
A unique capability of Merqury is its assessment of phased diploid assemblies. When parental k-mer sets are available, it can evaluate haplotype-specific completeness, phase block continuity, and switch errors, providing an invaluable resource for diploid genome projects [62] [63].
The following diagram illustrates the core workflow and logical relationship between the different assessment approaches:
Performance Comparison in Assembler Benchmarking
Recent benchmarking studies provide quantitative data on the performance of QUAST and Merqury in real-world scenarios, highlighting their complementary roles.
A 2025 benchmarking of hybrid de novo assembly tools for human and non-human data utilized both QUAST and Merqury alongside BUSCO to evaluate 11 different assembly pipelines. The study found that the Flye assembler, particularly when used with Ratatosk error-corrected long reads, outperformed others. The assessment showed that polishing significantly improved assembly quality, with two rounds of Racon followed by Pilon polishing yielding the best results as measured by these tools [33] [27].
Another study focusing on the repetitive yeast genome (Debaryomyces hansenii) assembled with four different sequencing platforms (PacBio Sequel, ONT MinION, Illumina NovaSeq 6000, and MGI DNBSEQ-T7) and seven assembly programs provided insights into platform-specific strengths. Oxford Nanopore with R7.3 flow cells generated more continuous assemblies than PacBio Sequel, despite some homopolymer-based errors. For short-read platforms, Illumina NovaSeq 6000 provided more accurate and continuous assembly, while MGI DNBSEQ-T7 offered a cheaper alternative for the polishing process [31].
The table below summarizes key quantitative findings from these benchmarking studies:
Table 1: Performance Metrics from Assembly Benchmarking Studies
| Assembler / Pipeline | QV (Merqury) | BUSCO % (QUAST) | N50 (QUAST) | Key Findings |
|---|---|---|---|---|
| Flye + Racon/Pilon | ~40-50 [33] | >95% [33] | Highest reported [33] | Best overall performance in hybrid assembly [33] |
| NextDenovo | N/A | Near-complete [21] | Single-contig assemblies [21] | Low misassemblies, stable across preprocessing [21] |
| Canu | N/A | High [21] | Fragmented (3-5 contigs) [21] | High accuracy but long runtimes [21] |
| ONT R7.3 | N/A | N/A | More continuous than PacBio [31] | Homopolymer errors but fewer chimeric contigs [31] |
Table 2: Analysis of Sequencing Platform Performance
| Sequencing Platform | Optimal Use Case | Error Profile | Assembly Continuity |
|---|---|---|---|
| PacBio Sequel | General long-read assembly | Less sensitive to GC content [31] | High but less than ONT R7.3 [31] |
| ONT MinION | Cost-effective continuous assemblies | Homopolymer-based indel errors [31] | Most continuous [31] |
| Illumina NovaSeq | SGS-first assembly or polishing | High accuracy, substitution errors [31] | Accurate and continuous for SGS [31] |
| MGI DNBSEQ-T7 | Cost-effective polishing | Accurate reads [31] | Cheap and accurate for polishing [31] |
Experimental Protocols for Tool Application
Protocol for QUAST Assessment
The standard workflow for QUAST involves the following steps:
- Input Preparation: Obtain the assembled contigs in FASTA format and a high-quality reference genome for the target organism.
- Tool Execution: Run QUAST with basic parameters. For a more comprehensive analysis, enable the
--gene-findingoption to assess gene space completeness and--buscofor universal single-copy ortholog assessment. - Output Interpretation: Analyze the generated
report.txtandreport.htmlfiles. Key metrics to examine include NG50 (contiguity relative to reference genome), total aligned length (completeness), and the number of misassemblies (correctness). Misassemblies are identified as breaks in the alignment to the reference and can indicate large-scale errors.
Protocol for Merqury Assessment
Merqury requires a different input preparation strategy, focusing on k-mer sets:
- K-mer Database Creation: First, build a k-mer database from high-accuracy Illumina reads using the
merylutility. The choice of k-mer size (k) is critical; a typical value is 21. - Tool Execution: Run Merqury with the k-mer database and the assembly to be evaluated.
- Output Interpretation: The key outputs include:
- QV and Completeness Scores: Found in
output_prefix.qualityandoutput_prefix.completeness. - Spectrum Plots: The
output_prefix.spectra-cn.pngfile provides a visual assessment. A "clean" plot where k-mer copy numbers in the assembly match expectations from the read set indicates a high-quality assembly. K-mers found only in the read set suggest missing sequences in the assembly, while k-mers with higher copy numbers in the assembly indicate potential artificial duplications [62].
- QV and Completeness Scores: Found in
For haploid assemblies, the analysis is straightforward. For diploid assemblies, the process can be extended by providing parental k-mer sets, enabling Merqury to generate haplotype-specific metrics and phasing statistics [63].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key bioinformatics tools and data types essential for conducting comprehensive genome assembly quality assessment.
Table 3: Essential Resources for Genome Assembly Assessment
| Tool / Resource | Category | Primary Function | Application Notes |
|---|---|---|---|
| Merqury | Software Tool | Reference-free quality assessment via k-mer comparison | Ideal for non-model organisms and phased diploid assembly evaluation [62] [63] |
| QUAST | Software Tool | Reference-based assembly quality assessment | Provides structural contiguity and misassembly metrics; requires a reference genome [62] |
| Meryl | Utility Software | K-mer counting and set operations | Required to build k-mer databases for Merqury analysis [63] |
| High-Accuracy Short Reads | Data Input | Source for k-mer database (e.g., Illumina) | Should be from the same individual as the assembly for valid Merqury analysis [62] |
| BUSCO | Software Tool | Assessment of gene space completeness | Works by searching for universal single-copy orthologs; can be run within QUAST [62] [33] |
| Reference Genome | Data Input | Gold standard for comparison | Critical for QUAST; quality directly impacts assessment validity [62] |
QUAST and Merqury represent two complementary paradigms for genome assembly assessment. QUAST excels in providing detailed structural insights when a high-quality reference is available, while Merqury offers a powerful reference-free approach that is particularly valuable for non-model organisms and for evaluating haplotype phasing in diploid genomes.
Evidence from recent benchmarking studies indicates that a combined approach, utilizing both tools, provides the most comprehensive evaluation. For instance, the best-performing pipelines, such as Flye with iterative polishing, were validated using both QUAST and Merqury metrics [33]. The choice between them—or the decision to use both—should be guided by the biological question, the availability of a reference genome, and the specific goals of the genomic study. As assembly methods continue to advance, the integration of multiple validation approaches will be essential for generating and verifying high-quality genome assemblies for biomedical and biological research.
Benchmarking Universal Single-Copy Orthologs (BUSCO) for Completeness Analysis
Within the critical process of benchmarking genome assemblers, the assessment of gene space completeness is a fundamental metric for evaluating the quality and utility of a genome assembly. Benchmarking Universal Single-Copy Orthologs (BUSCO) provides a standardized approach for this assessment, based on evolutionarily informed expectations of gene content from near-universal single-copy orthologs [64] [65]. This method offers a complementary metric to technical assembly statistics like N50, providing a biologically meaningful measure of completeness that enables robust comparisons across different assemblies and studies [66] [21]. This guide provides an objective comparison of BUSCO's performance against emerging alternatives, detailing its underlying methodology and integration within broader genome assembly benchmarking workflows.
The BUSCO Framework and Assessment Methodology
Core Principles and Workflow
The BUSCO evaluation system operates on a foundational principle: that all genomes within a specific lineage should share a core set of single-copy orthologous genes. These genes are evolutionarily conserved and are typically present as single copies, making them ideal markers for assessing genome completeness [64]. The assessment workflow involves comparing a genome assembly against curated datasets from OrthoDB, which contain hundreds to thousands of these conserved gene groups from various species [67] [66].
During analysis, BUSCO classifies genes into four distinct categories, providing a nuanced view of assembly quality [68] [64]:
- Complete (C): The gene sequence has been found in its entirety within the assembly.
- Single-Copy (S): The complete gene is present exactly once.
- Duplicated (D): The complete gene is present in multiple copies.
- Fragmented (F): Only a portion of the gene sequence has been identified.
- Missing (M): The gene sequence is entirely absent from the assembly.
The following diagram illustrates the standard BUSCO assessment workflow:
Standard Experimental Protocol
Implementing a BUSCO analysis requires careful attention to methodological parameters to ensure reproducible and accurate results. The following protocol outlines the standard procedure for conducting a BUSCO assessment:
Input Preparation: Obtain the genome assembly in FASTA format. The assembly can be at the contig, scaffold, or chromosome level [66].
Lineage Selection: Choose the appropriate BUSCO lineage dataset that matches the taxonomic group of the organism being analyzed. This is a critical step, as using an inappropriate lineage can lead to inaccurate results. Available lineages span major phylogenetic clades including Bacteria, Archaea, Eukaryota, Protists, Fungi, and Plants [64]. The lineage dataset can be specified using the
-lparameter.Analysis Mode Specification: Set the analysis mode using the
-mparameter based on the input data type [66]:genome: For DNA sequence assemblies (default mode)protein: For annotated protein sequencestranscriptome: For transcriptome assemblies
Computational Resources: Configure the number of parallel threads/cores using the
-cparameter to optimize runtime based on available computational resources [66].Execution: Run BUSCO with the specified parameters. The software will automatically download the necessary lineage dataset if not already present locally.
Output Interpretation: Analyze the generated results, including the short summary file, which provides the percentage of complete, duplicated, fragmented, and missing BUSCOs, along with visualizations such as pie charts for quick assessment [64].
Performance Comparison: BUSCO vs. Compleasm
Accuracy and Runtime Benchmarking
Recent developments in genome completeness assessment have introduced compleasm, a tool designed to address limitations in BUSCO's performance. As a reimplementation of BUSCO's core logic, compleasm utilizes the miniprot protein-to-genome aligner and conserved orthologous genes from BUSCO, claiming significant improvements in both speed and accuracy [67].
Experimental comparisons conducted across seven model organism reference genomes reveal notable performance differences between these tools. The table below summarizes a comprehensive benchmark analysis performed using standardized computational resources and datasets:
Table 1: Performance Comparison of BUSCO and Compleasm on Model Organism Reference Genomes
| Model Organism | Lineage Dataset | Tool | Complete (%) | Single-Copy (%) | Duplicated (%) | Fragmented (%) | Missing (%) | Total Genes (n) |
|---|---|---|---|---|---|---|---|---|
| H. sapiens (T2T-CHM13) | primates_odb10 | compleasm | 99.6 | 98.9 | 0.7 | 0.3 | 0.1 | 13,780 |
| BUSCO | 95.7 | 94.1 | 1.6 | 1.1 | 3.2 | 13,780 | ||
| M. musculus | glires_odb10 | compleasm | 99.7 | 97.8 | 1.9 | 0.3 | 0.0 | 13,798 |
| BUSCO | 96.5 | 93.6 | 2.9 | 0.6 | 2.9 | 13,798 | ||
| A. thaliana | brassicales_odb10 | compleasm | 99.9 | 98.9 | 1.0 | 0.1 | 0.0 | 4,596 |
| BUSCO | 99.2 | 97.9 | 1.3 | 0.1 | 0.7 | 4,596 | ||
| Z. mays | liliopsida_odb10 | compleasm | 96.7 | 82.2 | 14.5 | 3.0 | 0.3 | 3,236 |
| BUSCO | 93.8 | 79.2 | 14.6 | 5.3 | 0.9 | 3,236 | ||
| D. melanogaster | diptera_odb10 | compleasm | 99.7 | 99.4 | 0.3 | 0.2 | 0.1 | 3,285 |
| BUSCO | 98.6 | 98.4 | 0.2 | 0.5 | 0.9 | 3,285 |
The benchmark data reveals that compleasm consistently reports higher completeness percentages across most model organisms, with particularly significant differences observed for human (99.6% vs. 95.7%) and mouse (99.7% vs. 96.5%) genomes [67]. For the telomere-to-telomere (T2T) CHM13 human assembly, BUSCO reported a completeness of only 95.7%, whereas compleasm reported 99.6%, which aligns more closely with the annotation completeness of 99.5% [67].
In terms of computational efficiency, compleasm demonstrates substantial improvements, reportedly running 14 times faster than BUSCO for human genome assemblies [67]. This performance enhancement is particularly valuable when processing large genome assemblies or when conducting batch analyses of multiple genomes.
Methodological Differences Underlying Performance Variations
The performance disparities between BUSCO and compleasm stem from fundamental differences in their alignment strategies and processing workflows:
Alignment Algorithms: BUSCO employs MetaEuk for protein-to-genome alignment, typically running two rounds with different parameters to achieve sufficient sensitivity. In contrast, compleasm utilizes miniprot, a faster aligner that accurately detects splice junctions and frameshifts while performing only a single alignment round [67].
Orthology Confirmation: Both tools use HMMER3 to confirm orthology and filter out paralogous gene matches, retaining only matches above score cutoffs defined in lineage files [67].
Gene Representation: For each single-copy gene group with multiple protein sequences, both tools select the protein sequence with the highest HMMER search score to represent the group [67].
The combination of a more efficient alignment algorithm and streamlined workflow contributes to compleasm's superior speed performance while maintaining high accuracy.
BUSCO in Genome Assembler Benchmarking
Integration in Assembly Evaluation Pipelines
In comprehensive genome assembler benchmarking studies, BUSCO serves as an essential component of multi-faceted evaluation pipelines. These pipelines typically combine BUSCO with other assessment tools like QUAST (which provides technical metrics such as N50, contig count, and misassembly identification) to deliver a holistic view of assembly quality [21] [64].
A recent benchmark of eleven long-read assemblers using standardized computational resources exemplifies this approach. The study evaluated assemblies based on runtime, contiguity metrics (N50, total length, contig count), GC content, and completeness using BUSCO [21]. Results demonstrated that assemblers employing progressive error correction with consensus refinement, notably NextDenovo and NECAT, consistently generated near-complete, single-contig assemblies with high BUSCO completeness scores. Flye offered a strong balance of accuracy and contiguity, while Canu achieved high accuracy but produced more fragmented assemblies (3-5 contigs) with longer runtimes [21].
Table 2: BUSCO Results in Recent Genome Assembly Studies Across Various Species
| Study Organism | Sequencing Technology | Assembler Used | BUSCO Lineage | Complete (%) | Single-Copy (%) | Duplicated (%) | Fragmented (%) | Missing (%) |
|---|---|---|---|---|---|---|---|---|
| Butuo Black Sheep [69] | PacBio HiFi | Hifiasm | mammalia_odb10 | 95.9 | 93.5 | 2.4 | 1.1 | 3.0 |
| Human HG002 [27] | Nanopore + Illumina | Flye + Ratatosk | primates_odb10 | N/A | N/A | N/A | N/A | N/A |
| Multiple Prokaryotes [21] | Long-read | NextDenovo | bacteria_odb10 | >99* | N/A | N/A | N/A | N/A |
| Multiple Prokaryotes [21] | Long-read | NECAT | bacteria_odb10 | >99* | N/A | N/A | N/A | N/A |
*Note: Exact percentages not provided in source; described as "near-complete" assemblies.
Interpretation Guidelines for Assembly Quality
Proper interpretation of BUSCO results is crucial for accurate assessment of genome assemblies. The following guidelines assist researchers in diagnosing potential assembly issues based on BUSCO output patterns [64]:
High Complete, Low Duplicated/Fragmented/Missing: This ideal pattern indicates a well-assembled genome where core conserved genes are present in their entirety, suggesting the assembly is relatively accurate and captures most expected gene content.
High Duplicated BUSCOs: Elevated duplication rates may indicate assembly issues such as over-assembly, contamination, or unresolved heterozygosity, where alleles are assembled as separate sequences. This is particularly concerning in organisms not expected to have many paralogs or gene duplications.
High Fragmented BUSCOs: A high percentage of fragmented genes suggests assembly lacks continuity, potentially due to insufficient read length, low sequencing coverage, or assembly errors. This pattern often appears in repeat-rich regions that are challenging to assemble.
High Missing BUSCOs: Significant numbers of missing BUSCOs indicate substantial gaps in the assembly, potentially resulting from low sequencing coverage, assembly errors, or sequencing bias that underrepresents certain genomic regions.
Limitations and Evolutionary Considerations
While BUSCO provides valuable metrics for assembly completeness, recent research highlights important limitations and evolutionary considerations that affect result interpretation:
Taxonomic and Evolutionary Biases
Analysis of 11,098 eukaryotic genome assemblies from NCBI revealed that BUSCO gene content is significantly influenced by evolutionary history [70]. The study identified 215 taxonomic groups (out of 2,606 tested) that significantly varied from their respective lineages in terms of BUSCO completeness, while 169 groups displayed elevated complements of duplicated orthologs, likely resulting from ancestral whole genome duplication events [70].
Plant lineages showed a much higher mean BUSCO duplication rate (16.57%) compared to fungi (2.79%) and animals (2.21%), reflecting their different evolutionary histories and propensity for polyploidization [70]. These findings emphasize that deviations from "ideal" BUSCO scores may sometimes reflect biological reality rather than assembly quality issues.
Undetected Gene Loss and CUSCOs
A significant limitation of standard BUSCO analysis is its inability to account for undetected, yet pervasive, gene loss events across evolutionary lineages. One study estimated that between 2.25% to 13.33% of lineage-wise gene identifications may be misinterpreted using default BUSCO search parameters due to unaccounted gene loss [70].
To address this issue, researchers have developed a Curated set of BUSCO orthologs (CUSCOs) that provides up to 6.99% fewer false positives compared to standard searches across ten major eukaryotic lineages [70]. Additionally, syntenic BUSCO metrics offer higher contrast and better resolution for comparing closely related assemblies than standard BUSCO gene searches [70].
Table 3: Key Bioinformatics Tools and Resources for Genome Completeness Assessment
| Tool/Resource | Primary Function | Application Context | Key Features/Benefits |
|---|---|---|---|
| BUSCO [66] [64] | Genome completeness assessment | Evaluation of genome assemblies, gene sets, and transcriptomes | Evolutionarily informed expectations; standardized metric; multiple lineage datasets |
| Compleasm [67] | Genome completeness assessment | Faster alternative for large genomes or batch processing | Miniprot aligner; 14x faster than BUSCO; higher reported accuracy |
| QUAST [21] [64] | Assembly quality assessment | Technical evaluation of assembly contiguity and accuracy | Contiguity metrics (N50, L50); misassembly detection; reference-based comparison |
| OrthoDB [66] [70] | Ortholog database | Source of curated orthologous groups for BUSCO sets | Broad taxonomic sampling; functional and evolutionary annotations |
| HMMER [67] | Sequence homology search | Orthology confirmation in BUSCO/compleasm | Profile hidden Markov models for sensitive sequence detection |
| Miniprot [67] | Protein-to-genome alignment | Core aligner for compleasm | Fast splicing-aware alignment; accurate splice junction detection |
| MetaEuk [67] [66] | Protein-to-genome alignment | Core aligner for BUSCO (default mode) | Sensitivity to divergent sequences; reference-based gene discovery |
BUSCO remains an established standard for assessing genome completeness in assembler benchmarking, providing crucial biological context to complement technical metrics. Recent developments, particularly the introduction of compleasm, address significant limitations in runtime and accuracy while maintaining the core principles of conserved ortholog assessment. The integration of BUSCO metrics within comprehensive evaluation pipelines, coupled with appropriate interpretation that considers evolutionary histories, enables researchers to make informed decisions about assembly quality and suitability for downstream biological applications. As genome sequencing technologies continue to advance, completeness assessment tools will remain essential components of the genomics toolkit, with ongoing refinements improving their accuracy, efficiency, and biological relevance.
The quality of a de novo genome assembly is a cornerstone for downstream comparative and functional genomic studies, influencing the accuracy of variant identification, gene annotation, and evolutionary analysis [71] [27]. However, the assembly process is inherently challenging, especially for complex eukaryotic genomes replete with repetitive sequences [72] [73]. While metrics like contig N50 and scaffold N50 have traditionally been used to estimate assembly continuity, they can be misleading if long contigs are the result of misassemblies rather than accurate reconstruction [72] [74]. Similarly, gene space completeness metrics like BUSCO (Benchmarking Universal Single-Copy Orthologs) are invaluable but often reveal little about the assembly quality of the repetitive, intergenic regions that comprise the majority of many plant and animal genomes [72] [71] [75].
A particularly pernicious class of assembly errors involves structural misassemblies. These can range from small-scale indels to large-scale structural errors, such as the misjoining of two unlinked genomic fragments, which can profoundly distort downstream analyses like synteny comparisons and phylogenetic studies [71] [74]. The evaluation of repetitive sequence space has lagged behind gene space assessment, creating a critical gap in assembly validation [72]. This guide objectively compares the LTR Assembly Index (LAI), a reference-free metric specifically designed to evaluate the assembly of repetitive regions, with other modern methods for detecting misassemblies, providing researchers with a framework for comprehensive assembly benchmarking.
Understanding the LTR Assembly Index (LAI)
Conceptual Foundation and Methodology
The LTR Assembly Index (LAI) is a reference-free genome metric that evaluates assembly continuity by leveraging the properties of LTR retrotransposons (LTR-RTs), which are the predominant interspersed repeats in most plant genomes [72] [76]. The fundamental premise of LAI is that a more continuous and complete genome assembly will allow for the identification of a greater number of intact LTR-RTs. These elements are challenging to assemble correctly with short-read technologies due to their length and repetitive nature, making them a robust proxy for overall assembly quality, particularly in repetitive regions [72] [73].
The calculation of LAI follows a structured, four-step process, which can be implemented using the freely available LTR_retriever software [72]:
- Candidate Identification: LTR retrotransposon candidates are identified from the genome assembly using tools like
LTRharvestandLTR_FINDER. - False Positive Filtering: High-confidence LTR-RTs with perfect micro-structures (e.g., terminal motifs and target site duplications) are filtered from the candidate pool using
LTR_retriever, resulting in a set of intact LTR retrotransposons. - Whole-Genome Annotation: The entire genome is annotated for total LTR-RT content, including both intact and fragmented elements.
- Index Calculation: The LAI is calculated using the formula LAI = (Intact LTR-RTs / Total LTR-RTs) × 100. After correcting for LTR-RT amplification dynamics, this value provides a standardized score [72].
For LAI to be a reliable metric, the genome must meet minimum repeat content thresholds: intact LTR-RTs should contribute at least 0.1% to the genome size, and total LTR-RTs should constitute at least 5% [72] [76].
LAI in Action: Experimental Validation
The utility of LAI has been demonstrated in numerous genomic studies, often in direct comparison with other sequencing and assembly techniques. A pivotal application is evaluating the improvement gained from long-read sequencing.
In the assembly of the maize inbred line NC358, LAI was used to benchmark assemblies generated from PacBio datasets of varying depth and read length [77]. The study revealed that assemblies with higher sequence depth and longer reads achieved significantly higher LAI scores, reflecting their superior ability to resolve complex repetitive regions. Furthermore, the integration of high-quality optical maps dramatically improved contiguity, even for fragmented base assemblies [77].
Another key study compared genomic sequences produced by various sequencing techniques and revealed a "significant gain of assembly continuity by using long-read-based techniques over short-read-based methods," a conclusion clearly supported by LAI scores [72]. This makes LAI particularly valuable for iterative assembly improvement and assembler selection, as it can quantify gains in repeat region assembly that are invisible to BUSCO [72] [76].
The Broader Toolkit: Alternative Methods for Misassembly Detection
While LAI specializes in assessing the repeat space, a comprehensive assembly evaluation requires a multi-faceted approach. Several other tools have been developed to detect different types of assembly errors, ranging from single-nucleotide inaccuracies to large-scale structural misjoins.
Table 1: Comparison of Genome Assembly Assessment Tools
| Tool Name | Assessment Approach | Primary Strengths | Key Limitations |
|---|---|---|---|
| LTR Assembly Index (LAI) [72] | Reference-free; evaluates continuity using LTR retrotransposons. | Independent of genome size and gene space; ideal for repetitive regions. | Requires minimum LTR-RT content; underperforms in precise error calling [71]. |
| CRAQ [71] | Reference-free; uses clipped read alignment from raw reads. | Identifies errors at single-nucleotide resolution; distinguishes heterozygous sites from errors; pinpoints misjoin breakpoints. | Performance can be reduced in repeat regions with low read mapping [71]. |
| Merqury [71] [75] | Reference-free; based on k-mer comparisons between reads and assembly. | Provides single base error estimates; does not require a reference genome. | Cannot distinguish between base errors and structural errors [71]. |
| QUAST [71] [75] | Reference-based; compares assembly to a known reference. | Comprehensive reporting of misassemblies and structural differences. | Requires a closely related reference genome; misassemblies may be confused with genetic variation [71] [75]. |
| BUSCO [72] [75] | Reference-free; assesses presence/absence of conserved orthologous genes. | Excellent for evaluating gene space completeness and assembly completeness. | Does not assess repetitive, intergenic regions; can be inaccurate in polyploid genomes [72] [71]. |
| Inspector [71] | Reference-free; classifies assembly errors by scale. | Effective at detecting small-scale errors and regional collapses. | Has low recall for large-scale structural errors (CSEs) [71]. |
| CloseRead [75] | Reference-free; uses read alignment for targeted region validation. | Ideal for complex, polymorphic regions like immunoglobulin loci; provides intuitive visualizations. | More specialized for diagnosing specific problematic loci. |
Specialized Tools for Precision Error Detection
- CRAQ (Clipping information for Revealing Assembly Quality): This reference-free tool maps raw sequencing reads back to the assembled sequence to identify regional and structural errors based on clipped alignments [71]. Clipped reads—where only a portion of a read aligns to the assembly—often indicate large structural assembly errors, such as misjoins. CRAQ can distinguish these from heterozygous sites or haplotype structural differences and can transform error counts into Assembly Quality Indexes (AQIs) for regional (R-AQI) and structural (S-AQI) quality [71]. In benchmarking, CRAQ achieved an F1 score of over 97% for simulated errors, outperforming other reference-free tools like Inspector and Merqury in identifying structural errors [71].
- Merqury: This k-mer based method evaluates assembly accuracy by comparing the k-mers present in the original high-accuracy sequencing reads to those in the assembled sequence [71] [75]. While it provides excellent single-base error estimates and is fully reference-free, a key limitation is its inability to differentiate between single-nucleotide errors and larger structural errors [71].
- CloseRead: A recently developed approach for assessing local assembly quality in structurally complex and repeat-rich regions, such as immunoglobulin (IG) loci [75]. It aligns sequencing reads to the assembly and identifies errors like mismatches and coverage breaks, providing user-friendly visualizations. This is particularly useful for confirming the accurate assembly of biologically critical but difficult-to-assemble gene families [75].
Integrated Benchmarking: How the Tools Compare
To provide a holistic view of assembly quality, researchers should employ an integrated benchmarking strategy. The following diagram illustrates the relationship between different assessment tools and the genomic features they evaluate.
A compelling example of integrated benchmarking comes from a simulation study that compared several tools [71]. The reference-based QUAST-LG achieved the highest F1 score (>98%), as expected when a perfect reference is available. Among reference-free tools, CRAQ achieved the highest accuracy (F1 >97%) for detecting both Clip-based Regional Errors (CREs) and Clip-based Structural Errors (CSEs). Inspector showed good performance for CREs (~96% F1) but low recall for CSEs (28%). Merqury, while useful, had a lower aggregate F1 score of 87.7% [71]. This data highlights that tool selection should be guided by the specific types of errors a researcher aims to identify.
To implement the experimental protocols cited in this guide, researchers require access to specific data types and software tools.
Table 2: Essential Reagents and Resources for Assembly Evaluation
| Item Name | Type | Critical Function in Evaluation |
|---|---|---|
| Long-Read Sequencing Data (PacBio, Nanopore) | Data | Provides the long-range information necessary to span repeats and correctly assemble complex regions, which is crucial for achieving high LAI and low misassembly rates [73] [77]. |
| Short-Read Sequencing Data (Illumina) | Data | Serves as high-accuracy data for polishing long-read assemblies and is used by tools like CRAQ and Merqury for error detection [71] [14]. |
| LTR_retriever | Software | The core program required for accurate identification of intact LTR-RTs and subsequent LAI calculation [72]. |
| CRAQ | Software | Used for pinpointing assembly errors at single-nucleotide resolution and identifying precise breakpoints for structural misassemblies [71]. |
| Merqury | Software | Provides a fast, k-mer based evaluation of consensus quality (QV) and can flag problematic regions in the assembly [71] [75]. |
| Bionano Optical Maps | Data | An independent long-range mapping technology used to validate and correct large-scale scaffold structures, complementing sequence-based evaluation [77]. |
| Hi-C Data | Data | Used for chromosome-scale scaffolding and can also help validate large-scale structural assembly by confirming spatial contact patterns [71]. |
The benchmarking data clearly demonstrates that no single metric or tool provides a complete picture of genome assembly quality. The LTR Assembly Index (LAI) is an indispensable, reference-free metric for quantifying the assembly of repetitive sequences, a task for which traditional gene-completeness metrics are blind. Its independence from genome size and gene content makes it particularly valuable for plant genomes and other repeat-rich organisms.
However, LAI is not designed to identify the precise location of assembly errors or distinguish small-scale inaccuracies. For this, tools like CRAQ and Merqury are required. CRAQ excels in identifying the exact breakpoints of structural misassemblies, while Merqury provides a broad measure of base-level accuracy. Finally, BUSCO remains a critical first-pass check for gene space integrity.
Therefore, a robust genome assembly benchmarking protocol should be multi-layered:
- Use BUSCO to confirm core gene space completeness.
- Employ LAI to evaluate the assembly of repetitive regions, especially when using long-read technologies.
- Leverage CRAQ to identify and locate structural misassemblies and small-scale errors, particularly in regions of high biological interest.
- Utilize Merqury for a consensus quality check and to validate base-level accuracy.
This integrated approach ensures that both gene space and repeat space are accurately reconstructed, providing a solid foundation for all downstream genomic analyses.
The reconstruction of complete genome sequences from raw sequencing data is a cornerstone of modern genomics, enabling discoveries across evolutionary biology, disease research, and drug development. While long-read sequencing technologies from Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) have dramatically improved genome reconstruction, the choice of assembly software profoundly influences the quality and utility of the final output. Current benchmarking studies reveal that assemblers make distinct trade-offs between contiguity, base-level accuracy, and computational resource consumption. These trade-offs are not merely technical details but fundamentally influence the biological validity of downstream analyses in comparative genomics, variant discovery, and functional annotation. This guide synthesizes recent comprehensive benchmarking studies to objectively compare assembly tool performance, providing researchers with evidence-based recommendations for tool selection.
Performance Metrics and Benchmarking Methodology
Rigorous benchmarking requires standardized metrics and methodologies to ensure fair comparisons across diverse tools and datasets. The following sections outline the core evaluation criteria and experimental approaches used in contemporary assembly assessments.
Key Performance Indicators
- Contiguity: Typically measured by N50/NG50 (the length of the shortest contig/scaffold at 50% of the total assembly size) and the number of contigs. Higher N50 and lower contig counts indicate more complete reconstruction of chromosomal sequences.
- Completeness: Assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO), which quantifies the percentage of conserved, single-copy genes recovered in the assembly.
- Accuracy: Evaluated through base-level error rates and misassembly counts. Tools like QUAST and Merqury provide comprehensive quality assessments by comparing assemblies to reference genomes or k-mer spectra.
- Computational Efficiency: Measured by wall-clock time, CPU hours, and peak RAM usage.
Standardized Experimental Protocols
Benchmarking studies typically employ controlled computational environments with standardized datasets to ensure reproducible comparisons. A representative experimental workflow involves:
- Dataset Selection: Using well-characterized reference genomes (e.g., human HG002/NA24385) with available long-read and short-read data.
- Data Preprocessing: Applying uniform read filtering and correction across all tested assemblers.
- Assembly Execution: Running each assembler with optimized parameters on identical hardware.
- Quality Assessment: Applying the same battery of quality metrics (QUAST, BUSCO, Merqury) to all output assemblies.
- Resource Monitoring: Recording computational resources throughout the assembly process.
Comparative Performance of Genome Assemblers
Recent benchmarks of long-read assemblers reveal distinct performance profiles across multiple dimensions. The table below synthesizes key findings from comprehensive evaluations:
Table 1: Performance Comparison of Major Long-Read Assemblers
| Assembler | Best Application Context | Contiguity (Human N50) | BUSCO Completeness | Computational Demand (RAM) | Key Strengths |
|---|---|---|---|---|---|
| Flye | General-purpose; repetitive genomes | 26.6-38.8 Mbp [26] | 95.8% [78] | Moderate (329-502 GB) [26] | Excellent balance of accuracy and contiguity; robust across genomes [21] |
| NextDenovo | Large, repetitive, heterozygous genomes | High (specific values NA) | High (specific values NA) | Moderate | Superior for repetitive, heterozygous molluscan genomes [78] |
| GoldRush | Memory-constrained environments | 25.3-32.6 Mbp [26] | High (specific values NA) | Low (≤54.5 GB) [26] | Linear time complexity; efficient resource use [26] |
| Shasta | Rapid draft assemblies | 29.7-39.6 Mbp [26] | Moderate (requires polishing) [21] | Very High (885-1009 GB) [26] | Ultra-fast assembly; suitable for quick drafts [21] |
| Canu | Accuracy-focused projects | Lower (3-5 contigs) [21] | High [21] | Very High (specific values NA) | High accuracy; extensive error correction [21] |
| NECAT | Nanopore-specific assembly | High (specific values NA) | High (specific values NA) | Moderate | Progressive error correction; stable performance [21] |
Specialized assemblers have emerged for particular applications. For ancient metagenomic datasets characterized by ultrashort fragments and DNA damage patterns, CarpeDeam implements a damage-aware algorithm that outperforms conventional tools in recovering longer sequences from heavily degraded samples [79]. For hybrid assembly approaches combining long-read and short-read data, benchmarks demonstrate that Flye with Ratatosk-corrected long-reads followed by iterative polishing with Racon and Pilon produces optimal results [5].
The Assembly Trade-Off Triangle
The relationship between contiguity, accuracy, and computational resources forms a fundamental trade-off triangle in genome assembly. Benchmarks reveal that assemblers position themselves differently within this triangle:
- Contiguity vs. Accuracy: Assemblers like Shasta produce highly contiguous assemblies but often require additional polishing steps to achieve base-level accuracy comparable to Canu [21] [26]. Flye strikes a balance between these dimensions through its repeat graph approach [21].
- Accuracy vs. Resources: Canu achieves high accuracy through computationally intensive error correction, requiring substantially more time and memory than alternatives like GoldRush [21] [26].
- Contiguity vs. Resources: GoldRush challenges the conventional wisdom that high contiguity demands excessive resources by employing a novel dynamic data structure that foregoes all-versus-all read alignments [26].
Table 2: Trade-off Profiles of Major Assembler Types
| Assembler Category | Contiguity | Accuracy | Speed | Memory Efficiency |
|---|---|---|---|---|
| High-Resource OLC (Canu) | Medium | High | Low | Low |
| Balanced (Flye, NextDenovo) | High | High | Medium | Medium |
| Memory-Efficient (GoldRush) | High | Medium-High | Medium | High |
| Ultra-Fast (Shasta, Miniasm) | Medium | Low (requires polishing) | High | Medium |
Decision Framework for Assembler Selection
The following decision diagram illustrates the tool selection process based on project requirements and constraints:
Figure 1: Genome Assembler Selection Framework
Successful genome assembly projects require both computational tools and curated biological resources. The following table outlines essential components of the assembly toolkit:
Table 3: Essential Resources for Genome Assembly Projects
| Resource | Function | Examples/Specifications |
|---|---|---|
| Reference Genomes | Benchmarking and validation | GIAB HG002/NA24385 for human [5] |
| Sequence Read Archives | Source of experimental data | NCBI SRA, ENA [78] |
| Quality Assessment Tools | Assembly evaluation | QUAST, BUSCO, Merqury [26] [5] |
| Polishing Tools | Base-level error correction | Racon, Pilon [5] |
| Alignment Tools | Read-to-reference mapping | Minimap2 [80] [61] |
| Visualization Tools | Assembly inspection | Bandage, IGV [21] |
Contemporary benchmarking studies demonstrate that no single genome assembler achieves optimal performance across all metrics and applications. The choice between tools involves navigating key trade-offs between assembly contiguity, base-level accuracy, and computational resource requirements. Flye consistently delivers a balanced performance profile suitable for general-purpose assembly, while NextDenovo excels for complex, heterozygous genomes. For memory-constrained environments or large-scale projects, GoldRush offers an efficient alternative with linear time complexity. Specialized tools like CarpeDeam address unique challenges such as ancient DNA damage patterns. As sequencing technologies continue to evolve, ongoing benchmarking efforts will remain essential for guiding researchers toward appropriate tools that align with their specific scientific questions and resource constraints.
Conclusion
Benchmarking genome assemblers is not a one-size-fits-all process but a strategic exercise that balances contiguity, completeness, and correctness based on specific research goals. The choice of assembler and preprocessing steps jointly determines the final assembly quality, with progressive error correction tools like NextDenovo and NECAT often excelling in continuity, while Flye offers a strong balance of accuracy and contiguity. As we move into the telomere-to-telomere era, future advancements must address persistent challenges in assembling highly repetitive regions, complex polyploid genomes, and metagenomic samples. For biomedical research, adopting robust benchmarking practices is fundamental to generating reliable genomic resources that can power the discovery of disease mechanisms, drug targets, and clinically actionable variants, ultimately paving the way for more effective personalized medicine approaches.
References
- 1. An exploration of assembly strategies and quality metrics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8362059/]
- 2. Benchmark study for evaluating the quality of reference ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9988948/]
- 3. Methods and Tools for Assessing the Quality of Genome ... [https://www.cd-genomics.com/longseq/resource-assessing-the-quality-of-genome-assemblies.html]
- 4. GenomeQC: a quality assessment tool for genome assemblies ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6568-2]
- 5. Benchmarking of bioinformatics tools for the hybrid de novo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12284544/]
- 6. Functional annotation and comparative genomics analysis ... [https://www.nature.com/articles/s41598-023-41657-6]
- 7. Unlocking Microbial Secrets [https://www.numberanalytics.com/blog/comparative-genomics-microbial-genetics]
- 8. , Assembly , and Annotation in PATRIC, the... Comparative Genomics [https://pubmed.ncbi.nlm.nih.gov/29277864/]
- 9. Restauro-G: A Rapid Genome Re-Annotation System for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5054091/]
- 10. assessing the quality of genome assemblies with the 3 Cs [https://www.pacb.com/blog/beyond-contiguity/]
- 11. Toward a more holistic method of genome assembly ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-3382-4]
- 12. GAGE: A critical evaluation of genome assemblies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3290791/]
- 13. A proposed metric set for evaluation of genome assembly ... [https://www.sciencedirect.com/science/article/abs/pii/S0168952522002530]
- 14. Benchmarking short and long read polishing tools for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10582-x]
- 15. A comparative evaluation of genome assembly reconciliation ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1213-3]
- 16. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 17. Short read vs long read sequencing - INTEGRA Biosciences [https://www.integra-biosciences.com/united-states/en/blog/article/short-read-vs-long-read-sequencing]
- 18. Long-read human genome sequencing and its applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC7877196/]
- 19. A telomere-to-telomere genome assembly coupled with ... [https://www.nature.com/articles/s41588-025-02137-x]
- 20. Performance evaluation of six popular short-read simulators [https://www.nature.com/articles/s41437-022-00577-3]
- 21. Research Article Benchmarking long-read assembly tools ... [https://www.sciencedirect.com/science/article/pii/S2215017X2500058X]
- 22. Telomere-to-telomere gapless chromosomes of banana ... [https://www.nature.com/articles/s42003-021-02559-3]
- 23. Topsicle: a method for estimating telomere length from whole ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03783-4]
- 24. A telomere-to-telomere genome assembly coupled with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11985340/]
- 25. Efficient hybrid de novo assembly of human genomes with ... [https://www.nature.com/articles/s41587-020-00747-w]
- 26. Linear time complexity de novo long read genome ... [https://www.nature.com/articles/s41467-023-38716-x]
- 27. Benchmarking of bioinformatics tools for the hybrid de novo ... [https://pubmed.ncbi.nlm.nih.gov/40703096/]
- 28. Benchmarking hybrid assembly approaches for genomic ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-07041-8]
- 29. NextDenovo: an efficient error correction and accurate ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03252-4]
- 30. Systematic Comparison of the Performances of De Novo ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2021.696669/full]
- 31. A practical comparison of the next-generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9902641/]
- 32. Benchmarking seven most popular genome assemblers [https://medium.com/@shilparaopradeep/benchmarking-seven-most-popular-genome-assemblers-1eb249a94]
- 33. enchmarking of bioinformatics tools for the hybrid de novo ... [https://www.sciencedirect.com/science/article/pii/S2001037025002867]
- 34. Benchmarking of Hi-C tools for scaffolding plant genomes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11604747/]
- 35. Comparison of Hi-C-Based Scaffolding Tools on Plant ... [https://www.mdpi.com/2073-4425/14/12/2147]
- 36. Chromosome-level genome assembly and population ... [https://www.sciencedirect.com/science/article/pii/S2468265925001659]
- 37. Benchmarking of Hi-C tools for scaffolding plant genomes ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2024.1462923/full]
- 38. A complete diploid human genome benchmark for ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/41000953/]
- 39. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 40. Top five scientific trends shaping precision medicine in 2025 [https://sanogenetics.com/resources/blog/top-five-scientific-trends-shaping-precision-medicine-in-2025]
- 41. Assessing Read Quality, Trimming and Filtering - GitHub Pages [https://cloud-span.github.io/nerc-metagenomics03-qc-assembly/01-QC-quality-raw-reads/index.html]
- 42. Trimming and Filtering Data — RNA-Seq analysis of Mouse ... [https://cyverse-leptin-rna-seq-lesson-dev.readthedocs-hosted.com/en/latest/section-7.html]
- 43. Assembling Your DNA Sequences [https://www.geneious.com/guides/assembling-dna-sequences]
- 44. Comparison of long-read methods for sequencing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7751402/]
- 45. Benchmarking Long-Read Assemblers for Genomic Analyses of Bacterial Pathogens Using Oxford Nanopore Sequencing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7730629/]
- 46. Systematic Comparison of the Performances of De Novo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8415751/]
- 47. Comparative evaluation of Nanopore polishing tools for ... [https://www.nature.com/articles/s41598-021-00178-w]
- 48. Highly accurate genome polishing with DeepPolisher [https://research.google/blog/highly-accurate-genome-polishing-with-deeppolisher-enhancing-the-foundation-of-genomic-research/]
- 49. Benchmarking short and long read polishing tools for ... [https://pubmed.ncbi.nlm.nih.gov/38978005]
- 50. An open-source nanopore-only sequencing workflow for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12345217/]
- 51. Repetitive DNA sequence detection and its role in the ... [https://www.nature.com/articles/s42003-023-05322-y]
- 52. Assembly of highly repetitive genomes using short reads [https://pmc.ncbi.nlm.nih.gov/articles/PMC5989580/]
- 53. Haplotype-resolved assemblies and variant benchmark of a ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03116-3]
- 54. Haplotype-resolved diverse human genomes and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8026704/]
- 55. Faster and more affordable sequencing with the HiFi ... [https://www.pacb.com/blog/high-throughput-long-read-protocols/]
- 56. A haplotype-resolved chromosomal-level genome ... [https://www.nature.com/articles/s41597-025-05237-9]
- 57. Persistent memory as an effective alternative to random ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-05052-8]
- 58. Benchmarking workflows to assess performance and ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03934-3]
- 59. MEGAHIT v1.0: A fast and scalable metagenome ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202315301183]
- 60. SPAdes: A New Genome Assembly Algorithm and Its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3342519/]
- 61. Tradeoffs in alignment and assembly-based methods for ... [https://www.nature.com/articles/s41467-024-46614-z]
- 62. Merqury: reference-free quality, completeness, and phasing ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02134-9]
- 63. marbl/merqury: k-mer based assembly evaluation [https://github.com/marbl/merqury]
- 64. Genome Completeness Assessment with BUSCO [https://www.biobam.com/genome-completeness-assessment-with-busco/]
- 65. BUSCO - SIB Swiss Institute of Bioinformatics [https://www.expasy.org/resources/busco]
- 66. BUSCO - from QC to gene prediction and phylogenomics [https://busco.ezlab.org/]
- 67. compleasm: a faster and more accurate reimplementation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10558035/]
- 68. Benchmarking Universal Single-Copy Orthologs (BUSCO ... [https://www.ncbi.nlm.nih.gov/datasets/docs/v2/data-processing/policies-annotation/quality/busco/]
- 69. Genome assembly and whole-genome resequencing study ... [https://www.nature.com/articles/s41597-025-05805-z]
- 70. Universal orthologs infer deep phylogenies and improve ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02328-2]
- 71. Identification of errors in draft genome assemblies at single ... [https://www.nature.com/articles/s41467-023-42336-w]
- 72. Assessing genome assembly quality using the LTR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6265445/]
- 73. Impact of short-read sequencing on the misassembly of a ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-07397-5]
- 74. Genome assembly forensics: finding the elusive mis-assembly [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2008-9-3-r55]
- 75. CloseRead: a tool for assessing assembly errors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12090573/]
- 76. Assessing genome assembly quality using the LTR ... [https://www.pacb.com/publications/assessing-genome-assembly-quality-using-the-ltr-assembly-index-lai/]
- 77. Effect of sequence depth and length in long-read assembly ... [https://www.nature.com/articles/s41467-020-16037-7]
- 78. Benchmarking Oxford Nanopore read assemblers for high- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8059532/]
- 79. CarpeDeam: a de novo metagenome assembler for heavily ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03839-5]
- 80. A comprehensive benchmarking of adaptive sampling tools for ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03729-w]