Complete Guide to ONT Ultra-Long Read Assembly: From Fundamentals to Clinical Applications in 2024
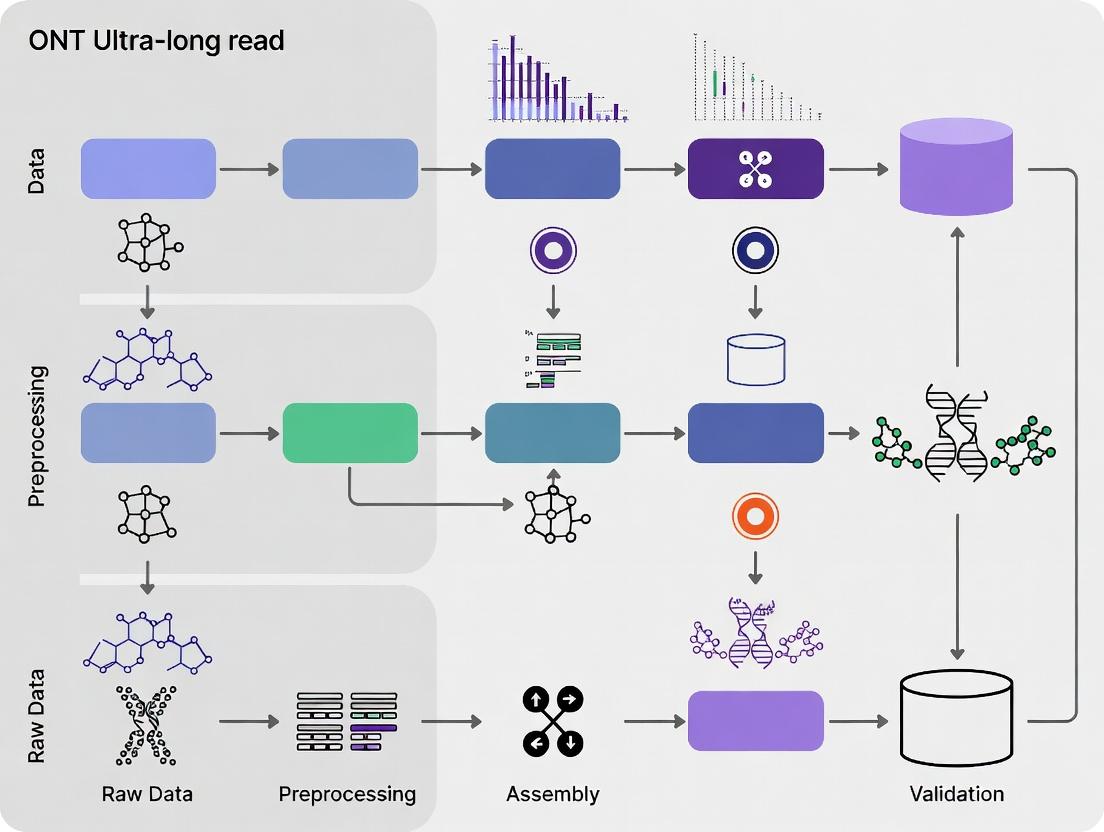
This comprehensive guide provides researchers and drug development professionals with a detailed workflow for Oxford Nanopore Technologies (ONT) ultra-long read assembly.
Complete Guide to ONT Ultra-Long Read Assembly: From Fundamentals to Clinical Applications in 2024
Abstract
This comprehensive guide provides researchers and drug development professionals with a detailed workflow for Oxford Nanopore Technologies (ONT) ultra-long read assembly. It explores the foundational principles of ultra-long read sequencing, presents step-by-step methodological pipelines from sample preparation to polished assembly, addresses common troubleshooting and optimization challenges, and validates results through comparative analysis with short-read and hybrid methods. The article concludes by highlighting the transformative impact of complete genome assemblies on biomedical research, including structural variant discovery, epigenetic characterization, and clinical diagnostics.
What is ONT Ultra-Long Read Sequencing? Core Concepts and Advantages for Modern Genomics
Within the broader thesis on optimizing Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows, a precise and quantitative definition of "ultra-long reads" is paramount. This application note clarifies the core metrics—read length distributions, N50, and L50—used to characterize ultra-long sequencing datasets, which are critical for achieving high-quality, contiguous genome assemblies in research and drug development.
Core Metrics Defined
- Read Length Distribution: The frequency of reads across different length bins. Ultra-long protocols shift this distribution significantly toward longer lengths.
- N50 Read Length: The length of the shortest read in the set of longest reads that together represent 50% of the total bases sequenced. It is a weighted median statistic.
- L50 Count: The minimum number of reads whose summed length equals 50% of the total assembly length or total bases sequenced.
Table 1: Representative Metrics from Contemporary ONT Ultra-Long Sequencing Studies
| Study / Sample | Mean Read Length (kb) | N50 Read Length (kb) | Longest Read (kb) | Total Yield (Gb) | Protocol Key Feature |
|---|---|---|---|---|---|
| Human HG002 (UL Kit 10.1) | ~60 | ~95 | >800 | ~60 | Ligation-based UL sequencing |
| Arabidopsis (v14 chemistry) | ~70 | ~115 | >1,000 | ~40 | R10.4.1 flow cell, high input mass |
| Typical "Standard" Read Dataset | 10 - 30 | 15 - 40 | 100 - 200 | >20 | Standard Ligation Kit (SQK-LSK114) |
Experimental Protocol: Assessing Ultra-Long Read Metrics
Protocol 1: Generating and Evaluating Ultra-Long Read Datasets
Objective: To generate an ultra-long read library from high molecular weight (HMW) genomic DNA and calculate key length distribution metrics.
Materials & Reagents (Research Toolkit)
| Item | Function |
|---|---|
| HMW gDNA (>50 kb) | Starting material; integrity is critical for ultra-long reads. |
| ONT Ultra-Long DNA Sequencing Kit (SQK-ULK114) | Contains specialized reagents for minimal DNA fragmentation. |
| R10.4.1 or R10.4.1 flow cell | Pore version optimized for high-accuracy, long reads. |
| PippinHT or BluePippin System | For precise size selection of >50 kb fragments. |
| Qubit Fluorometer & dsDNA HS Assay | Accurate quantification of low-concentration HMW DNA. |
| Nanopore Sequencing Device (PromethION/GridION) | Platform for running the sequencing experiment. |
| Guppy (v6.4.6+) or Dorado basecaller | Converts raw electrical signals to nucleotide sequences (FASTQ). |
| NanoPlot (v1.41.0) | Tool for creating read length distribution plots and summary stats. |
| SeqKit (v2.6.0) | Lightweight tool for FASTA/Q file manipulation and stat calculation. |
Methodology:
- DNA Extraction & QC: Isolate gDNA using a gentle method (e.g., Nanobind HMW kit). Assess integrity via pulsed-field gel electrophoresis (PFGE) or FEMTO Pulse; target average size >100 kb.
- Library Preparation: Follow the ONT Ultra-Long protocol. Key steps involve:
- Minimal DNA Repair & End-Prep: Limited incubation time.
- No Fragmentation: Omit standard bead-based shearing steps.
- Adaptor Ligation: Use UL-specific adaptors with reduced incubation.
- Size Selection (Optional but Recommended): Use a PippinHT system to enrich for fragments >50 kb.
- Sequencing: Load library onto a fresh R10.4.1 flow cell. Start a PromethION 48h sequencing run with active loading to maximize data output.
- Basecalling & Demultiplexing: Use the super-accuracy (sup) model in Dorado basecaller (live or post-run) to generate FASTQ files.
- Metric Calculation:
- Run NanoPlot:
NanoPlot --fastq reads.fastq.gz --loglength -o nanoplot_results --N50 - This generates a summary statistic file (
NanoStats.txt) and a read length distribution plot. - Calculate N50/L50 Manually (Alternative): a. Sort all reads by length (longest to shortest). b. Calculate the total sum of base pairs (B). c. Cumulatively sum lengths from the longest read downward. d. The L50 is the number of reads at which the cumulative sum first exceeds B/2. e. The N50 is the length of the shortest read in this set.
- Run NanoPlot:
Workflow and Conceptual Diagrams
Title: Ultra-Long Read Generation & Analysis Workflow
Title: Conceptual Diagram of N50 and L50 Calculation
This Application Note details the fundamental principles of Oxford Nanopore Technologies (ONT) sequencing, from the biophysics of the nanopore to the computational process of basecalling. The information is framed within the context of a broader thesis research project focused on optimizing ultra-long read assembly workflows for de novo genome assembly and structural variant detection. Understanding the core technology is essential for researchers, scientists, and drug development professionals to effectively design experiments, troubleshoot protocols, and interpret data derived from nanopore sequencing platforms.
Nanopore Chemistry and Sensing Principle
At the heart of ONT sequencing is a charged, protein nanopore (e.g., CsgG) embedded within an electrically resistant polymer membrane. An ionic current is established by applying a voltage across the membrane. As a DNA or RNA molecule is processively threaded through the pore via a motor protein, the distinct chemical groups of each nucleotide (A, C, G, T, U) cause characteristic disruptions in the ionic current. These disruptions are not binary signals for individual bases but are complex "squiggles" representing ~5-6 nucleotides within the pore constriction at any given time.
Table 1: Key Nanopore System Components and Their Functions
| Component | Material/Example | Primary Function in Sequencing |
|---|---|---|
| Membrane | Artificial polymer (e.g., proprietary) | Provides a stable, insulating layer to house the nanopore and sustain an ionic gradient. |
| Nanopore | Protein complex (e.g., R10.4.1, R9.4.1) | Forms a transmembrane channel for DNA translocation. The internal structure dictates signal sensitivity. |
| Motor Protein | Helicase (DNA) or DSP (Direct RNA) | Controls the rate and direction of DNA/RNA translocation through the pore. |
| Buffer | High-concentration electrolyte (e.g., LiCl, KCl) | Conducts ionic current. Composition affects current noise and signal quality. |
| Sensor Chip | Application-Specific Integrated Circuit (ASIC) | Contains thousands of individual sensor wells, each capable of measuring picoampere-scale current changes. |
Signal Acquisition to Sequence: The Basecalling Workflow
The raw signal (current over time) must be converted into a DNA/RNA sequence. This process, known as basecalling, is a computational challenge solved using machine learning models.
Diagram Title: Nanopore Signal to Sequence Basecalling Pipeline
Table 2: Evolution of ONT Basecalling Models and Accuracy (Representative Data)
| Basecaller Model Type | Key Characteristics | Approximate Single-Read Accuracy* | Best For |
|---|---|---|---|
| Hidden Markov Model (HMM) | Early, statistical models (Albacore). | ~92% (R9.4) | Historical data analysis. |
| Recurrent Neural Network (RNN) | Flip-flop models (Guppy v3-v5). | ~95-97% (R9.4) | Balanced speed & accuracy. |
| CRISPR-Cas9 Enhanced | Uses guide RNAs for modification detection. | N/A (for 5mC, 5hmC) | Direct epigenetic calling. |
| High-Accuracy Models (Q20+) | Newer architectures (Bonito, Dorado). | >99% (R10.4.1, duplex) | Ultra-long read assembly, variant detection. |
*Accuracy is chemistry- and context-dependent. R10.4.1 and duplex sequencing significantly improve accuracy.
Protocol: Conducting a Standard Ultra-Long DNA Sequencing Run
This protocol outlines the key steps for preparing and running an ultra-long DNA sequencing library on a PromethION device, a common platform for large-scale assembly projects.
Objective: Generate ultra-long (>100 kbp) reads from high molecular weight (HMW) genomic DNA for de novo genome assembly. Materials: See "The Scientist's Toolkit" below.
Procedure:
Part A: DNA Quality Assessment and Repair
- Quantify and Quality Check: Use a Qubit fluorometer for concentration and a Femto Pulse or pulsed-field gel electrophoresis system to assess DNA fragment size distribution. Target a modal length >50 kbp.
- DNA Repair: In a 0.2 mL PCR tube, combine:
- 1-5 µg HMW gDNA (in 45 µL TE).
- 7 µL NEBNext FFPE DNA Repair Buffer.
- 3 µL NEBNext FFPE DNA Repair Mix.
- 5 µL Ultra II End-prep reaction buffer.
- 2 µL Ultra II End-prep enzyme mix.
- Mix gently, spin down, and incubate at 20°C for 15 minutes, then 65°C for 15 minutes. Immediately place on ice.
Part B: Adapter Ligation and Bead-Based Cleanup
- Prepare Adapter Mix: To the 62 µL repaired DNA, add:
- 25 µL Blunt/TA Ligation Master Mix.
- 10 µL of the Ligation Sequencing Adapter (AMX).
- 5 µL of the Ultra-Long Sequencing Adapter (ULA).
- Mix thoroughly by pipetting. Incubate at room temperature for 30 minutes.
- Cleanup: Add 100 µL of AMPure XP beads (0.4x ratio) to the 102 µL ligation mix. Mix and incubate for 5 minutes. Pellet beads, wash twice with 70% ethanol, and air-dry for 30 seconds.
- Elute the purified library in 25 µL of Elution Buffer (EB). Transfer to a new tube.
Part C: Priming and Loading the Flow Cell
- Flow Cell Priming: Uncap the FLO-PRO002M (PromethION) flow cell. Inject 800 µL of Priming Mix (FLP) into the priming port at a steady rate. Wait 5 minutes.
- Prepare Loading Mix: In a fresh tube, combine:
- 25 µL of the purified library.
- 12.5 µL Sequencing Buffer (SQB).
- 37.5 µL Loading Beads II (LBII). Mix by pipetting gently.
- Load the Library: Open the SpotON sample port. Add 75 µL of the Loading Mix dropwise to the port. Close the port.
- Begin Sequencing: Insert the flow cell into the PromethION device. Start the sequencing run via MinKNOW software, selecting the appropriate "Ultra-Long" sequencing script.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for ONT Ultra-Long Sequencing
| Item (Example Kit) | Function in Workflow |
|---|---|
| Ultra-Long DNA Sequencing Kit (SQK-ULK114) | Provides specialized enzymes and buffers for end-prep, ligation of ultra-long adapters, and motor protein loading. |
| Ligation Sequencing Adapter (AMX) | Short, tether adapters that bind the motor protein to the DNA fragment, enabling controlled translocation. |
| Ultra-Long Sequencing Adapter (ULA) | Specialized adapter that promotes the loading of extremely long DNA molecules into pores. |
| Flow Cell Priming Kit (EXP-FLP002) | Contains the priming buffer (FLP) required to wet and prepare the flow cell's internal channels prior to loading the library. |
| AMPure XP Beads | Magnetic beads used for size selection and cleanup of DNA libraries. Ratios (e.g., 0.4x, 0.8x) control size cutoff. |
| NEBNext FFPE DNA Repair Mix | Enzyme mix for repairing nicks, gaps, and deaminated bases common in HMW DNA, crucial for read length. |
| Qubit dsDNA HS Assay Kit | Fluorometric quantitation specific for double-stranded DNA, essential for accurate input measurement. |
| MinION/PromethION Flow Cell (R10.4.1) | The consumable sensor device containing the nanopore array. R10.4.1 pores offer improved homopolymer accuracy. |
Data Analysis Considerations for Assembly Workflows
For ultra-long read assembly, the choice of basecaller and subsequent filters is critical. The workflow typically involves generating raw reads, basecalling, quality filtering, and assembly.
Diagram Title: Ultra-Long Read Assembly Analysis Workflow
Application Notes
Within the context of a broader thesis on Oxford Nanopore Technologies (ONT) ultra-long read assembly workflow research, three key advantages define its transformative impact on genomics. Ultra-long reads (>100 kb, with reports exceeding 4 Mb) enable the resolution of complex genomic landscapes that are intractable to short-read technologies.
1. Spanning Repeats: Long tandem repeats, segmental duplications, and transposable elements collapse or misassemble in short-read assemblies. ONT reads can completely span these regions, anchoring unique flanking sequences and accurately resolving repeat length and structure. This is critical for studying telomeres, centromeres, and disease-associated repeat expansions (e.g., in FMR1, C9orf72).
2. Phasing Haplotypes: Ultra-long reads preserve long-range allelic information, enabling the separation of maternal and paternal chromosomes over multi-megabase distances—entire chromosome arms. This allows for the construction of fully phased diploid assemblies, revealing cis-regulatory interactions and compound heterozygosity in Mendelian disorders.
3. Detecting Structural Variants (SVs): ONT reads provide direct, single-molecule evidence for large-scale genomic alterations (>50 bp), including deletions, duplications, inversions, translocations, and complex rearrangements. The long read length increases the probability of capturing both breakpoints within a single read, enabling precise mapping and typing of SVs, which are a major contributor to genetic diversity and disease.
Table 1: Performance Metrics of ONT Ultra-Long Read Workflows in Genomic Studies
| Metric | Typical Range (Ultra-long Protocols) | Comparison to Short-Read NGS | Key Impact |
|---|---|---|---|
| Read Length (N50) | 50 kb - >100 kb | 150-300 bp | Spans most repetitive elements |
| Max Read Length | Up to 4 Mb reported | ~600 bp | Enables telomere-to-telomere assembly |
| Phasing Block N50 | 10 - 100 Mb | < 1 Mb | Haplotype resolution across entire genes/chromosomes |
| SV Detection Sensitivity | >95% for >1 kb variants | < 30% for >1 kb variants | Comprehensive variant catalog |
| Repeat Resolution | Directly spans repeats up to read length | Collapses repeats longer than read length | Accurate assembly of complex regions |
Table 2: Common Structural Variants Detected by ONT Ultra-Long Reads
| SV Type | Size Range | Detection Mechanism | Relevance in Disease |
|---|---|---|---|
| Deletion | 50 bp - >1 Mb | Direct read alignment gap | Tumor suppressor loss, genetic disorders |
| Insertion | 50 bp - >1 Mb | Novel sequence within aligned read | Drug resistance genes, novel sequences |
| Inversion | >1 kb - Mb | Split-read with inverted alignment | Developmental disorders, gene disruption |
| Duplication | >1 kb - Mb | Increased read coverage & split alignment | Gene dosage diseases (e.g., Charcot-Marie-Tooth) |
| Translocation | N/A | Reads aligning to two different chromosomes | Cancer driver events, fusion genes |
Experimental Protocols
Protocol 1: Ultra-Long DNA Extraction and Library Preparation for ONT Sequencing
Objective: To generate high molecular weight (HMW) DNA (>150 kb) suitable for ultra-long read sequencing on platforms like the PromethION. Materials: Fresh tissue or cells, Nuclei isolation buffer, Nanobind HMW DNA Extraction Kit (Circulomics), Magnetic separator, Qubit fluorometer, Broad Range dsDNA assay, Pulse-field gel electrophoresis (PFGE) system. Procedure:
- Nuclei Isolation: Homogenize tissue/cells in cold nuclei isolation buffer. Centrifuge to pellet nuclei.
- HMW DNA Extraction: Resuspend nuclei pellet and proceed with Nanobind disk-based extraction per manufacturer's protocol, with gentle mixings. Elute in low-EDTA TE buffer.
- DNA QC: Quantify using Qubit. Assess size distribution using PFGE or Genomic DNA ScreenTape. Aim for a modal size >50 kb.
- Library Preparation: Use the Ligation Sequencing Kit (SQK-LSK114). Minimize pipetting and vortexing. Use ½ reaction volumes for all enzymatic steps to conserve sample. Do not shear DNA.
- Loading: Load library onto a primed R10.4.1 flow cell. Sequence for up to 72 hours with active loading replenishment.
Protocol 2: De Novo Assembly and Phasing using Shasta and HapDuplex
Objective: To generate a fully phased, diploid de novo assembly from ultra-long reads. Software: Shasta assembler, HapDuplex (for assembly graph-based phasing), Verkko pipeline (optional), Minimap2, HiGlass for visualization. Procedure:
- Basecalling & QC: Basecall raw FAST5 files using Super Accurate model (
dorado basecaller). Filter reads by length (e.g.,--min-length 50000). - Shasta Assembly: Run Shasta with
--inputas filtered reads. Use configNanopore-UL. This produces an initial assembly graph. - HapDuplex Phasing: Run HapDuplex on the Shasta assembly graph to partition reads into two haplotype-specific sets.
- Haplotype-Specific Assembly: Re-assemble each read set separately using Shasta, producing two haplotype-resolved assemblies (haplotype 1, haplotype 2).
- Polishing: Align all ultra-long reads to each haplotype assembly with Minimap2. Polish using Racon (4-5 iterations) followed by Medaka.
- Evaluation: Assess completeness with BUSCO, phasing continuity with phase block N50, and consensus accuracy with Mercury.
Protocol 3: Structural Variant Calling with Sniffles2
Objective: To detect and genotype SVs from aligned ultra-long reads. Software: Minimap2, Sniffles2, IGV or pggb for visualization. Procedure:
- Alignment: Align ultra-long reads to a reference genome (e.g., GRCh38) using Minimap2 with preset
map-ont(-ax map-ont). - Sort & Index: Sort and index the BAM file using
samtools sortandsamtools index. - SV Calling: Run Sniffles2:
sniffles --input aligned.sorted.bam --reference ref.fa --vcf output.vcf --minsvlen 50. For population/genotype calling, use the--snfintermediate file and joint calling mode. - Filtering: Filter VCF based on SUPPORT reads, genotype quality (GQ), and variant quality (QUAL). Consider using
--minsupport 5. - Annotation & Visualization: Annotate SVs with gene overlap using SnpEff or AnnotSV. Load BAM and VCF into IGV to inspect read evidence for high-priority SVs.
Visualization
ONT UL Workflow from Sample to Analysis
Long Reads Span Repeats vs Short Read Collapse
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for ONT Ultra-Long Read Workflows
| Item Name | Supplier/Example | Function in Workflow |
|---|---|---|
| Nanobind HMW DNA Kit | Circulomics / PacBio | Gentle, disk-based extraction preserving ultra-high molecular weight DNA integrity. |
| Ligation Sequencing Kit (LSK) | Oxford Nanopore | Prepares DNA libraries by attaching sequencing adapters without PCR, maintaining read length. |
| R10.4.1 Flow Cell | Oxford Nanopore | Pore version providing higher raw accuracy, crucial for SNP and small variant calling within long reads. |
| ProNex Size-Selective Beads | Promega / Beckman Coulter | Precise size selection to enrich for ultra-long fragments prior to library prep. |
| Low-EDTA TE Buffer | Various | Elution/storage buffer that minimizes DNA degradation and chelation of Mg²⁺ needed for sequencing enzymes. |
| Pulse-Field Gel Electrophoresis Ladder | Bio-Rad / NEB | High-range molecular weight standard for accurately assessing DNA fragment sizes >50 kb. |
| Critical Dry Ice / Cold Blocks | Various | Maintaining samples at cold temperatures during all steps to inhibit nuclease activity. |
Within the broader thesis on optimizing Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows, the selection of sequencing hardware and chemistry is paramount. The transition from the R9.4.1 to the R10.4.1 flow cell, paired with the SQK-LSK114 ligation sequencing kit, represents a significant advancement for generating high-accuracy, ultra-long reads. This combination addresses key challenges in de novo genome assembly, haplotype phasing, and structural variant detection in complex genomic regions, which are critical for genetic disease research and therapeutic target identification.
Quantitative Comparison of Key Components
Table 1: Flow Cell Characteristics (R9.4.1 vs. R10.4.1)
| Feature | R9.4.1 Pore | R10.4.1 Pore |
|---|---|---|
| Pore Structure | Single constriction | Dual reader head (two sensing regions) |
| Nominal Accuracy (1D) | ~94-96% | ~97-99% (Q20+ mode available) |
| Key Improvement | Established technology | Enhanced homopolymer resolution (5-mer sensing) |
| Optimal Read Length | All lengths | Superior for Ultra-Long Reads (>100 kb) |
| Primary Benefit for UL Assembly | Longer historical data | Higher per-read accuracy improves assembly continuity |
Table 2: Sequencing Kit Comparison (SQK-LSK109 vs. SQK-LSK114)
| Feature | SQK-LSK109 (R9.4.1) | SQK-LSK114 (R10.4.1) |
|---|---|---|
| Compatible Flow Cell | R9.4.1 | R10.4.1 (Flongle, MinION, PromethION) |
| Recommended DNA Input | 1 µg (no fragmentation) | 1-3 µg (no fragmentation for UL) |
| Library Prep Time | ~60-90 minutes | ~75 minutes |
| Key Chemistry | Ligation-based | Ligation-based with V14 Sequencing Chemistry |
| Critical for UL Workflow | Supports UL reads | Optimized for R10.4.1, enabling Q20+ and duplex modes |
Detailed Experimental Protocols
Protocol 1: Ultra-Long DNA Extraction & Quality Assessment for R10.4.1/LSK114 Objective: To obtain high molecular weight (HMW) DNA (>150 kb N50) suitable for ultra-long sequencing.
- Cell Lysis: Use gentle, non-mechanical lysis (e.g., agarose plug lysis for cultured cells or modified CTAB for tissue). Avoid vortexing or vigorous pipetting.
- DNA Purification: Employ size-selective magnetic bead-based cleanups (e.g., SPRI beads) with reduced binding time to retain long fragments. Alternatively, use pulsed-field gel electrophoresis for size selection.
- Quantification & QC: Use fluorometric assays (Qubit HS DNA kit). Assess fragment size distribution via pulsed-field gel electrophoresis or FEMTO Pulse system. Target an N50 > 50 kb, ideally > 100 kb.
- DNA Repair: If necessary, use a gentle DNA repair enzyme mix, followed by AMPure XP bead cleanup (0.4x ratio).
Protocol 2: Library Preparation with SQK-LSK114 Kit for Ultra-Long Reads Note: Perform all steps in a PCR-free clean environment with low-binding tips.
- End-Prep & dA-Tailing: Combine 1-3 µg HMW DNA with NEBNext Ultra II End-prep buffer and enzyme. Incubate at 20°C for 5 minutes, then 65°C for 5 minutes. Clean up with 0.4x AMPure XP beads. Elute in 25 µL EB.
- Adapter Ligation: Add 25 µL of LNB (Ligation Buffer), 5 µL of T4 DNA Ligase, and 5 µL of AMX (Adapter Mix) directly to the eluate. Mix gently and incubate at room temperature for 20 minutes.
- Adapter-Bead Binding & Elution: Add 50 µL of LFB (Library Binding Buffer) and transfer to a tube containing pre-washed FAB (Flow Cell Adapter Beads). Resuspend gently and incubate for 5 minutes. Pellet beads, remove supernatant, and wash twice with 125 µL LWB (Library Wash Buffer). Elute DNA library in 15 µL EB for 10 minutes.
- Priming & Loading the R10.4.1 Flow Cell: a. Prime the flow cell: Mix 800 µL of FLP (Flow Cell Priming Buffer) with 200 µL of nuclease-free water. Load 200 µL of this mix into the flow cell via the priming port. b. Prepare the sequencing mix: Combine 12 µL of SQB (Sequencing Buffer), 8.5 µL of LLB (Library Loading Beads), and 11.5 µL of the eluted library. c. Load the sequencing mix dropwise to the spot on the flow cell. Close the priming port and begin the "Platform QC" script, followed by the "High Accuracy (UC)" or "Super Accuracy (SUP)" script for optimal basecalling.
Visualization of Workflow and Logic
Title: UL Sequencing Workflow for Genome Assembly
Title: Pore Evolution Impact on Assembly
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for ONT Ultra-Long Read Workflow
| Item | Function in Workflow |
|---|---|
| Magnetic Beads (SPRI/AMPure XP) | Size-selective purification and cleanup of HMW DNA and libraries. Critical for retaining ultra-long fragments. |
| Low-Bind/Non-Stick Microcentrifuge Tubes & Tips | Minimizes DNA shearing and surface adhesion loss of precious HMW samples during all steps. |
| Pulsed-Field Gel Electrophoresis (PFGE) System | Gold-standard for visualizing and assessing the size distribution of HMW DNA (N50, N90). |
| Qubit Fluorometer with HS DNA Kit | Accurate quantification of low-concentration DNA samples without degradation from intercalating dyes. |
| NEBnext Ultra II End Prep Module | Component of LSK114 kit; performs DNA end repair and dA-tailing for adapter ligation. |
| Flow Cell Adapter Beads (FAB) | Magnetic beads in LSK114 kit that specifically bind adapter-ligated DNA for purification. |
| Library Loading Beads (LLB) | Reagent in LSK114 kit that increases library density for optimal loading onto the flow cell. |
| nuclease-free Water (PCR Grade) | Used in all dilution and elution steps to prevent enzymatic degradation of the library. |
Application Notes
Within the broader research on Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows, three target applications demonstrate the transformative impact of continuous, multi-megabase reads. These applications overcome limitations inherent to short-read and hybrid assembly approaches.
- De Novo Assembly: Ultra-long reads dramatically increase contiguity, producing fewer, more complete contigs. This reduces the complexity of scaffolding and minimizes gaps, enabling high-quality draft assemblies from a single sequencing technology.
- Telomere-to-Telomere (T2T) Projects: Achieving truly complete, gapless chromosomes requires spanning long, repetitive regions such as centromeric satellite arrays, ribosomal DNA (rDNA) clusters, and segmental duplications. ONT ultra-long reads are the primary data type for de novo assembly of these regions in flagship projects like the T2T Consortium's CHM13 human genome.
- Complex Genomic Regions: This includes medically relevant loci with high GC content, long tandem repeats, or complex structural variations (SVs). Examples are the PCDH gene clusters, the MUC gene family, and the Major Histocompatibility Complex (MHC). Ultra-long reads allow for the complete phasing and resolution of such regions.
Table 1: Quantitative Impact of Ultra-Long Reads in Recent Studies (2023-2024)
| Study / Project Focus | Key Metric | Result with Standard Reads (N50) | Result with Ultra-Long Reads (N50) | Improvement Factor |
|---|---|---|---|---|
| Human T2T Assembly (CHM13) | Contig Continuity (Chromosome X) | ~50 Mb (CLR) | Full arm (~155 Mb) | >3x |
| Plant Genome (Hexaploid Wheat) | Assembly Contiguity | 1.2 Mb | 22.5 Mb | ~19x |
| Complex SV Resolution in Cancer | Median Size of Precisely Resolved SVs | < 1 kb | > 50 kb | >50x |
| Bacterial Assembly (Repeat-Rich) | Number of Contigs | 105 | 1 (complete circularized) | 105x reduction |
Protocols
Protocol 1: Ultra-Long DNA Extraction and Size Selection for T2T Projects
Objective: Isolate high molecular weight (HMW) DNA with fragments >150 kb, with a significant fraction >1 Mb, suitable for T2T assembly.
- Cell Lysis: Gently lyse cells embedded in low-melt agarose plugs or using a liquid-phase protocol with minimal pipetting (e.g., Circulomics Nanobind HMW DNA Kit).
- RNase A/Proteinase K Digestion: Incubate lysate at 50°C for 60 minutes.
- Magnetic Bead Clean-up: Use a 2:1 ratio of solid-phase reversible immobilization (SPRI) beads to sample for initial short-fragment removal. Retain supernatant.
- DNA Precipitation: Precipitate HMW DNA using isopropanol. Use a wide-bore pipette tip to spool the DNA.
- Size Selection (Blue Pippin): Load DNA onto a 0.75% agarose cassette in a Blue Pippin or PippinHT system. Set size cut-off to 150 kb. Elute in low-EDTA TE buffer.
- Quality Control: Assess yield via Qubit HS dsDNA assay. Assess size distribution via FEMTO Pulse or genomic DNA ScreenTape analysis. Target A260/A280 ~1.8 and A260/A230 >2.0.
Protocol 2: ONT Ligation Sequencing for Ultra-Long Reads (SQK-LSK114)
Objective: Prepare an ultra-long read sequencing library with minimal fragmentation.
- DNA Repair and End-Prep: Incubate 3 µg of size-selected HMW DNA with NEBNext FFPE DNA Repair Mix and Ultra II End-prep enzyme mix for 30 minutes at 20°C, then 30 minutes at 65°C. Clean with 0.4x SPRI beads.
- Native Barcode Ligation (Optional): For multiplexing, ligate Native Barcode Adapters (EXP-NBD114) using NEB Blunt/TA Ligase for 30 minutes at room temperature (RT). Pool barcoded samples and clean with 0.4x SPRI beads.
- Adapter Ligation: Ligate Sequencing Adapters (AMII) to the DNA using NEB Quick T4 DNA Ligase for 30 minutes at RT. Use a 0.2x SPRI bead clean-up to retain the largest fragments. Critical: Do not over-clean.
- Priming and Loading: Mix Sequencing Buffer (SQB II) and Loading Beads (LB II). Add the library and load onto a primed R10.4.1 or R10.4.1 flow cell.
- Sequencing: Run on a GridION or PromethION for up to 72 hours with active channel selection enabled.
Protocol 3:De NovoAssembly and Polishing Workflow
Objective: Assemble and polish a high-contiguity genome from ultra-long reads.
- Basecalling & QC: Perform high-accuracy basecalling with
dorado(>=v0.5.0) using thesupmodel. Assess read length distribution withNanoPlot. - Assembly: Assemble using
flye(>=v2.9) with--nano-hqmode orshasta(for human) with appropriate--inputlength parameters. - Polishing:
- Polish 1 (Racon/Medaka): Use
medaka_consensuswith the appropriate model (e.g.,r1041_e82_400bps_sup_v5.0.0). - Polish 2 (Short-Read Polish): If available, align Illumina reads to the assembly using
bwa mem. Call variants withclair3orbcftools mpileupand apply them usingbcftools consensus.
- Polish 1 (Racon/Medaka): Use
- Evaluation: Compute assembly metrics with
QUAST. Assess completeness withBUSCO.
Diagrams
Diagram 1: UL Assembly Workflow for T2T Genomes
Diagram 2: Resolving Complex Genomic Regions
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Reagent | Function in Ultra-Long Workflow |
|---|---|
| Nanobind HMW DNA Kit (Circulomics) | Liquid-phase extraction minimizing shear, yielding >100 kb DNA. |
| Megaruptor 3 System (Diagenode) | Programmable DNA shearing; used for controlled reduction of DNA size if required for library prep. |
| Blue Pippin / PippinHT System (Sage Science) | Automated, precise size selection via pulsed-field electrophoresis in agarose gel cassette. |
| Femto Pulse System (Agilent) | Capillary electrophoresis for accurate sizing and quantification of ultra-long DNA fragments (>165 kb). |
| Ligation Sequencing Kit SQK-LSK114 (ONT) | Optimized library prep chemistry for ultra-long reads, minimizing DNA damage. |
| R10.4.1 Flow Cell (ONT) | Nanopore with a dual reader head, providing very high (>Q20) raw accuracy for homopolymers and repeats. |
| NEBNext FFPE DNA Repair Mix (NEB) | Repairs nicks and base damage in HMW DNA, critical for maximizing read length. |
| SPRIselect Beads (Beckman Coulter) | Solid-phase reversible immobilization beads for precise, low-shear clean-up and size selection. |
Step-by-Step ONT Ultra-Long Read Assembly Workflow: From DNA Extraction to Polished Contigs
The successful generation of ultra-long reads (>100 kbp) for Oxford Nanopore Technologies (ONT) sequencing is a cornerstone of de novo genome assembly projects, enabling the resolution of complex genomic regions, structural variants, and repetitive elements. The quality of the final assembly is intrinsically linked to the initial input DNA. This protocol details the best practices for High Molecular Weight (HMW) DNA extraction, quantification, and quality control (QC), framed within the context of an ONT ultra-long read assembly workflow thesis. Robust HMW DNA is the critical first step, upon which all subsequent library preparation, sequencing, and bioinformatic assembly efforts depend.
HMW DNA Extraction: Core Principles and Protocol
The primary goal is to isolate DNA with minimal mechanical and nuclease-induced shearing, preserving molecules longer than 150 kbp, with an ideal target of >50 kbp as a minimum for ultra-long protocols.
2.1 Key Principles:
- Minimize Physical Shear: Use wide-bore pipette tips, gentle mix by inversion, and avoid vortexing, vigorous pipetting, or rapid centrifugation of DNA solutions.
- Inhibit Nucleases: Use fresh, cold EDTA-containing buffers and protease/RNase treatments during lysis. Keep samples on ice when possible.
- Purify Effectively: Remove contaminants like proteins, lipids, polysaccharides, and short-fragment DNA that can inhibit downstream enzymes.
2.2 Detailed Protocol: Magnetic Bead-Based HMW DNA Cleanup (Post-Extraction)
This protocol follows a typical column- or bead-based extraction (e.g., Qiagen Genomic-tip, Monarch HMW DNA Extraction Kit) and details a final size-selective cleanup using SPRI (Solid Phase Reversible Immobilization) beads.
Materials:
- HMW DNA in elution buffer (e.g., TE, EB).
- Size-Selective SPRI Beads (e.g., Circulomics SRE, Pacific Biosciences SMRTbell beads).
- Fresh 70% and 80% Ethanol (in nuclease-free water).
- Wide-bore pipette tips (200 µL, 1000 µL).
- Low-bind microcentrifuge tubes or 1.5 mL LoBind tubes.
- Magnetic rack suitable for tube format.
Method:
- Equilibrate: Bring all reagents and samples to room temperature (RT) to prevent precipitation.
- Binding: Add a calculated volume of size-selective SPRI beads (typically at a 0.4-0.6x sample:bead ratio to bind and retain fragments >~15-30 kbp). Mix gently by slowly inverting the tube 10 times. Do not vortex.
- Incubate: Incubate at RT for 5-10 minutes. Place tube on magnetic rack until supernatant clears (2-5 minutes).
- Wash: Carefully remove and discard the supernatant without disturbing the bead pellet. With tube on magnet, add 500 µL of freshly prepared 80% ethanol. Incubate for 30 seconds, then remove and discard ethanol. Repeat with a second 500 µL wash of 80% ethanol. Ensure all ethanol is removed.
- Dry: Let the bead pellet air-dry on the magnet for 2-3 minutes. Do not over-dry.
- Elute: Remove tube from magnet. Add desired volume of elution buffer (e.g., 50-100 µL 10mM Tris-HCl, pH 8.0-8.5). Gently pipette-mix using a wide-bore tip. Incubate at RT for 5 minutes.
- Recover: Place tube back on magnet. Once cleared, carefully transfer the supernatant containing the purified HMW DNA to a new low-bind tube.
Quality Control: Fragment Analyzer and FEMTO Pulse
Accurate QC is non-negotiable. Agarose gel electrophoresis is insufficient. Capillary electrophoresis systems provide precise size distribution and quantification.
3.1 Detailed QC Protocol: Using the Agilent Femto Pulse System
The Femto Pulse system is optimized for very high sensitivity and large fragment analysis.
Reagents: Genomic DNA 165 kb Kit (Agilent, Part Number FP-1002). Sample Preparation:
- Prepare samples at 1-2 ng/µL in nuclease-free water or elution buffer. Overly concentrated DNA can saturate the signal.
- Prepare the marker (M1) by adding 40 µL of deionized water to the lyophilized pellet. Vortex thoroughly.
- Mix 5 µL of sample with 5 µL of marker (M1) in a PCR tube or microplate well. Mix by pipetting.
- Denature at 75°C for 5 minutes, then immediately place on ice for 5 minutes.
- Centrifuge briefly before loading.
Instrument Run:
- Prime the capillary array with gel and conditioning solution as per manufacturer instructions.
- Load the prepared samples into the sample plate.
- Set the run method to "Genomic DNA 165 kb."
- Initiate the run. Analysis typically takes ~90 minutes.
Data Interpretation: Key metrics are the Weighted Average (WA) Size (in kbp) and the Percentage of Fragments >50 kb or >150 kb. A high-quality HMW prep for ultra-long sequencing should have a WA >50 kbp and >30-40% of fragments >150 kbp.
3.2 Quantitative Data Summary
Table 1: QC Metric Benchmarks for ONT Ultra-Long Sequencing
| QC Metric | Minimum Requirement | Optimal Target | Instrument/Method |
|---|---|---|---|
| Concentration | >30 ng/µL | 50-100 ng/µL | Qubit dsDNA HS Assay |
| A260/A280 | 1.8 - 2.0 | 1.9 - 2.0 | Nanodrop (screen only) |
| Weighted Avg. Size | >30 kbp | >50 kbp | Fragment Analyzer / Femto Pulse |
| % of Fragments >50 kb | >50% | >70% | Fragment Analyzer / Femto Pulse |
| % of Fragments >150 kb | >20% | >40% | Femto Pulse |
Table 2: Comparison of Capillary Electrophoresis Systems for HMW DNA QC
| Feature | Fragment Analyzer (FA) | Femto Pulse |
|---|---|---|
| Optimal Size Range | Up to 60 kbp | Up to 165 kbp |
| Sample Sensitivity | ~0.5-5 ng/µL | 0.5 pg/µL - 50 ng/µL |
| Key Metric | Max detectable peak, DV50/200 | Weighted Average, %>150kb |
| Throughput | Higher (96-well) | Standard (48- or 96-well) |
| Best For | General HMW QC, plasmid analysis | Ultra-long DNA profiling, low input |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for HMW DNA Workflows
| Item | Function & Importance |
|---|---|
| Size-Selective SPRI Beads | Selective binding of long DNA fragments; crucial for removing short fragments that consume sequencing pores. |
| Wide-Bore/Low-Bind Pipette Tips | Minimizes physical shearing forces during liquid handling and reduces DNA adhesion to plastic surfaces. |
| High-EDTA Lysis Buffers | Chelates Mg2+ ions, inactivating Mg2+-dependent nucleases that degrade DNA during extraction. |
| RNAse A & Proteinase K | Degrades RNA and cellular proteins, yielding pure, protein-free DNA essential for clean library prep. |
| Qubit dsDNA HS Assay Kit | Fluorescence-based assay specific for double-stranded DNA; provides accurate concentration without contamination interference. |
| Femto Pulse Genomic DNA 165 kb Kit | Provides optimized gel matrix, markers, and conditions for precise sizing of ultra-large DNA fragments. |
Visualized Workflows
Title: HMW DNA Extraction & QC Decision Workflow
Title: Thesis Workflow: From HMW DNA to Genome Assembly
Application Notes and Protocols
Within the broader thesis on Oxford Nanopore Technologies (ONT) ultra-long (UL) read assembly workflow research, the library preparation stage is the critical bottleneck. The ultimate goal of generating N50 read lengths exceeding 100 kilobases (kb) is directly contingent on preserving native DNA fragment length and selectively enriching for the longest molecules. This protocol details a refined methodology for ultra-long DNA library preparation, emphasizing gentle handling and precise size selection.
1. Core Principles and Current Data Ultra-long read library preparation departs from standard protocols by prioritizing the avoidance of mechanical and enzymatic shearing. Key quantitative benchmarks from recent optimizations are summarized below.
Table 1: Comparative Impact of DNA Handling Methods on Fragment Integrity
| Handling Method | Average Fragment Size (kb) | N50 (kb) | Protocol Deviation from Standard |
|---|---|---|---|
| Standard Pipette Mixing | 15-30 | 40-60 | Vortexing & vigorous pipetting |
| Gentle Wide-Bore Pipetting | 80-120 | >150 | Using wide-bore tips, slow pipette actions |
| Needle Shearing (21G) | 10-20 | 30 | Intentional shearing for short-read protocols |
Table 2: Performance of Size Selection Methods for UL Reads
| Size Selection Method | Target Size Retention | Approximate Yield Loss | Key Application |
|---|---|---|---|
| Short Fragment Buffer (SFB) Wash | >10 kb | 30-50% | Quick cleanup; removes very short fragments. |
| Blue Pippin (Sage Science) with 0.75% Agarose Cassette | >50 kb | 60-80% | High-precision selection for UL libraries. |
| Automated (e.g., Covaris g-TUBE) | User-defined | 40-60% | More reproducible than manual shearing. |
2. Detailed Protocol for Ultra-Long DNA Library Preparation
Materials: High Molecular Weight (HMW) DNA (>50 kb N50), Ultra-Long Fragment Buffer (ONT SQK-ULK001 kit), Wide-bore pipette tips (200 µL, 1000 µL), Magnetic beads (Solid Phase Reversible Immobilization, SPRI), Blue Pippin system with 0.75% DF Marker S1 agarose cassette.
Part A: DNA Normalization and Repair (Minimizing Shearing)
- Quantification: Use fluorescence-based assays (e.g., Qubit dsDNA BR Assay). Avoid spectrophotometers (e.g., NanoDrop) due to contamination insensitivity.
- DNA Handling: Always pre-wet wide-bore tips. Mix reactions by slowly pipetting up and down 5-10 times. Do not vortex or spin tubes vigorously.
- End-prep & Repair: Combine 1-5 µg HMW DNA with Ultra-Long Fragment Buffer and enzyme mix. Incubate at 20°C for 20 minutes, then 65°C for 20 minutes. Use wide-bore tips throughout.
Part B: Size Selection via Blue Pippin
- Sample Preparation: Add recommended internal standards to the end-prepped DNA.
- Cassette Loading: Load sample into a 0.75% DF Marker S1 agarose cassette. Set the instrument to collect fragments >50 kb.
- Recovery: After size selection, recover DNA in a ~40 µL elution buffer. The yield will be low (nanogram quantities) but suitable for adapter ligation.
Part C: Adapter Ligation and Clean-up
- Ligation: Combine size-selected DNA with Adapter Mix (AMX) and Ligation Buffer (LNB). Incubate at room temperature for 30 minutes.
- Bead-based Clean-up: Use a double SPRI bead clean-up.
- First, add 0.4x volumes of SPRI beads to bind and remove excess adapters. Retain the supernatant containing large, adapter-ligated fragments.
- To the supernatant, add a further 0.8x volumes of SPRI beads (total 1.2x). Bind, wash, and elute the final library in 15 µL Elution Buffer (ELB).
3. Workflow and Decision Pathway Visualization
Diagram Title: Ultra-Long Read Library Prep Decision Workflow
Diagram Title: Double SPRI Bead Clean-up Process
4. The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Ultra-Long Read Library Prep
| Item | Function in Protocol | Critical Note |
|---|---|---|
| Wide-Bore Pipette Tips | Minimizes hydrodynamic shear during liquid transfer. | Must be used for all steps post-DNA extraction. |
| Solid Phase Reversible Immobilization (SPRI) Beads | Selective binding of DNA by size in polyethylene glycol (PEG) solutions. | Lower PEG/bead ratios retain longer fragments. |
| Blue Pippin System (Sage Science) | Automated, high-resolution size selection using pulsed-field electrophoresis. | 0.75% agarose cassettes are optimal for >50 kb fragments. |
| Qubit dsDNA BR Assay Kit | Accurate quantification of low-concentration, long DNA without degradation. | Preferable over absorbance methods for purity and sensitivity. |
| ONT SQK-ULK001 Kit | Optimized enzyme and buffer system for ultra-long DNA end-prep and ligation. | Formulated for minimal incubation times to reduce handling. |
| Low-Bind Microcentrifuge Tubes | Reduces DNA adhesion to tube walls, maximizing recovery. | Essential post-size selection where DNA mass is minimal. |
Within the research framework of ONT ultra-long read assembly workflows, optimizing sequencing run management is critical for generating the contiguous, high-quality data required for de novo genome assembly, structural variant detection, and epigenetic analysis. This protocol details a methodology for maximizing DNA yield and read length through integrated live basecalling and run monitoring. The approach focuses on real-time decision-making to extend the productive phase of a sequencing run, directly contributing to the generation of ultra-long reads.
The following quantitative parameters, derived from current ONT documentation and recent literature, are essential for live run management.
Table 1: Critical Metrics for Live Run Monitoring & Intervention
| Metric | Target Range/Value | Function & Rationale |
|---|---|---|
| Active Pores | >40% of loaded pores | Indicates sufficient available sequencing capacity. A sharp, continuous drop may signal DNA depletion or pore blockages. |
| Read Length N50 (Live) | Increasing trend; target >50 kb | Key indicator of ultra-long read success. Real-time tracking allows for assessment of library quality and run health. |
| Pore Speed (bases/sec) | Consistent, ~70-120 bps for R10.4.1 | Significant deviations can indicate voltage instability or motor protein issues. |
| Yield per Hour | Stable or increasing linear phase | Enables accurate prediction of total run yield. A plateau signals the run's end. |
| Read Count vs. Mean Read Length | Negative correlation is ideal | As the run progresses, an increase in mean length with a slowing of new starts indicates successful ultra-long sequencing. |
Table 2: Protocol Decision Matrix Based on Live Metrics
| Observed Issue (Live Metrics) | Potential Cause | Recommended Protocol Action |
|---|---|---|
| Rapid decline in active pores, short reads | DNA library depleted | Initiate in-run reload protocol (see below) to introduce fresh library. |
| High pore count but low yield/speed | Voltage or buffer instability | Check flow cell integrity; ensure no bubbles. Adjust voltage if within manufacturer specs. |
| Long reads but low N50 | DNA fragment nicks/breaks | Focus on pre-sequencing DNA extraction & repair. Continue run, but optimize next prep. |
| Yield plateau, pores still active | Motor protein/nucleotide limitation | Perform in-run flush with nuclease to clear stalled pores, followed by a reload. |
Experimental Protocol: In-Run Re-Load for Yield Extension
This protocol is triggered via live basecalling analysis when active pores fall below 30% while sequencing buffer remains.
Materials & Reagents:
- Fresh, prepped ultra-long DNA library (SQK-LSK114)
- Nuclease-free water
- Running Buffer with Fuel (RBF; ONT, EXP-FLP002)
- Flow Cell Wash Kit (WSH004)
- MinKNOW software (v22.12+)
- MinION or PromethION device
Procedure:
- Pause Sequencing: In MinKNOW, click "Pause" for the ongoing run. Select the "Stop for wash" option.
- Unload Flow Cell: Follow the software prompt to unload the flow cell from the sequencing position.
- Initial Wash:
- Draw off ~200 µL of buffer from the priming port using a pipette.
- Load 200 µL of fresh Running Buffer with Fuel into the flow cell via the priming port. Wait 5 minutes.
- Library Introduction:
- Prepare a fresh library mix: 12 µL fresh library + 18 µL RBF + 20.5 µL nuclease-free water.
- Draw off 30 µL from the priming port, then slowly load the 50.5 µL library mix dropwise.
- Ensure no bubbles are introduced.
- Resume Sequencing:
- Re-insert the flow cell into the device.
- In MinKNOW, select "Resume" and choose the existing run. Live basecalling will resume automatically.
- Post-Reload Monitoring:
- Monitor the "Active Pores" metric for recovery (target >40% within 30 mins).
- Verify that the "Read Length N50" continues its previous trajectory.
Visualizations
Diagram 1: Live Basecalling-Enabled Run Management Workflow
Diagram 2: Key Factors Influencing Ultra-Long Read Length
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Ultra-Long Read Sequencing & Live Management
| Item (Example Product) | Function in Workflow |
|---|---|
| Ultra-Long DNA Extraction Kit (Circulomics Nanobind / QIAGEN Genomic-tip) | Preserves multi-Mbp chromosomal DNA fragments, the foundational input for ultra-long reads. |
| DNA Damage Repair Mix (ONT SQK-LSK114 component) | Repairs nicks and breaks in high-MW DNA that would prematurely terminate reads. |
| High-Salt Library Buffer (ONT Ligation Sequencing Kit) | Enhards DNA compaction, promoting translocation of ultra-long fragments through nanopores. |
| Running Buffer with Fuel (ONT EXP-FLP002) | Maintains optimal pH, ionic strength, and provides energy (fuel) for the motor protein during sequencing. |
| Flow Cell Wash Kit (ONT WSH004) | Clears blocked pores (nuclease flush) and refreshes buffer system for run extension protocols. |
| Remora-based Mod Kit (e.g., Dorado duplex) | Enables real-time, high-accuracy basecalling and modification calling (5mC, 6mA), integral to live analysis. |
| MinKNOW Software | The core platform for controlling the sequencer, performing live basecalling, and providing real-time run metrics. |
Within the broader research on Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows, the primary data analysis steps of basecalling and initial quality assessment are critical. The transition from raw electrical signal (fast5) to nucleotide sequence (fastq) via sophisticated basecallers like Dorado directly impacts downstream assembly continuity and accuracy. Subsequent quality control with NanoPlot provides essential metrics to evaluate read suitability for ultra-long assembly, informing decisions on sequencing sufficiency and need for additional data generation. This protocol details the application of these tools in a production bioinformatics pipeline.
Application Notes: Dorado Basecalling
Dorado is a high-performance, CUDA-accelerated basecaller developed by Oxford Nanopore Technologies. It supersedes earlier tools like Guppy, offering significant speed improvements and continuous integration of the latest pore models and algorithms (e.g., duplex, modified base detection).
Key Features:
- Performance: Utilizes NVIDIA GPUs for real-time or batch basecalling.
- Models: Supports a variety of models tailored for specific kit ligations (SQK-LSK114, SQK-RBK114), accuracy modes (
high,super,fast), and applications (duplex, DNA/RNA, 5mC/6mA detection). - Output: Produces standard
fastqfiles and can emit additional data like modified base probabilities in.bamformat.
Quantitative Performance Data
Table 1: Comparative Basecalling Performance (Representative Data)
| Tool | Speed (samples/sec) | Typical Read Accuracy (Q-score) | GPU Memory Requirement | Key Output |
|---|---|---|---|---|
| Dorado (super-acc) | ~1800 | ~Q20 (98.99%) | 4-8 GB | fastq, .bam with mods |
| Guppy (HAC) | ~400 | ~Q18 (98.41%) | 2-4 GB | fastq |
| Dorado Duplex | ~50 | >Q25 (99.68%) | 8+ GB | Duplex fastq |
Detailed Protocol: Basecalling with Dorado
1. Prerequisite Setup
- Hardware: NVIDIA GPU (e.g., A100, V100, RTX 3090) with compatible drivers and CUDA >= 11.8.
- Software: Install Dorado via the provided download link from the ONT Community or using Conda:
conda create -n dorado -c bioconda dorado. - Data: ONT raw sequencing data in
fast5orpod5format (recommended).
2. Execute Basecalling Navigate to the directory containing the raw data and run Dorado. The basic command structure is:
Example command for high-accuracy basecalling of pod5 files:
For modified base detection (5mC) alongside basecalling:
3. Output Organization
The primary fastq file is ready for QC. The .bam file from modified base calling contains both sequence and methylation scores, viewable with tools like samtools.
Application Notes: Read QC with NanoPlot
NanoPlot generates comprehensive quality control summaries from ONT fastq files. It is essential for assessing read length distribution, average quality, and yield—key parameters for determining if the data meets the input requirements for ultra-long read assemblers like Shasta or Canu.
Key QC Metrics Reported
Table 2: Essential QC Metrics from NanoPlot for Ultra-Long Read Assembly
| Metric | Target for Ultra-Long Assembly | Interpretation |
|---|---|---|
| N50 Read Length | >50 kb (preferably >100 kb) | Indicator of long-read continuity. |
| Mean Read Quality (Q) | >Q15 | Low quality may necessitate filtering or re-basecalling. |
| Total Yield (Gb) | Dependent on genome size (e.g., >50X cov.) | Sufficient coverage for assembly. |
| Read Length Distribution | Long tail towards 100+ kb | Visual confirmation of ultra-long content. |
Detailed Protocol: Quality Assessment with NanoPlot
1. Installation Install NanoPlot via pip or conda:
2. Generate QC Report
Run NanoPlot on the basecalled fastq file:
3. Analyze Output
The tool generates an HTML summary report (NanoPlot-report.html) and numerous plots (.png). Critical files include:
NanoPlot-report.html: Interactive summary.LengthvsQualityScatterPlot_dot.png: Scatter plot of read length vs. average quality.Yield_By_Length.png: Cumulative yield plot.
4. Decision Point Based on the report, decide if the data is sufficient for assembly:
- Proceed: If N50 and yield meet targets.
- Filter: Use
filtlongorNanoFiltto remove short/low-quality reads. - Resequence: If yield is insufficient.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Components for ONT Primary Data Analysis Workflow
| Item | Function | Example/Note |
|---|---|---|
| ONT Sequencing Kit (Ligation) | Prepares genomic DNA for sequencing by adding motor proteins and adapters. | SQK-LSK114 for ultra-long reads. |
| Dorado Basecaller Software | Converts raw electrical signals (pod5) to nucleotide sequences (fastq). |
Requires NVIDIA GPU and license. |
| High-Performance Compute Node | Provides the computational resources for accelerated basecalling. | NVIDIA GPU (e.g., A100, V100), >=32 GB CPU RAM. |
| NanoPlot/NanoPack Suite | Generates visualizations and statistics for read QC. | Critical for assessing data pre-assembly. |
| Reference Genome (Optional) | Used for calculating read alignment identity metrics during QC. | e.g., CHM13 for human samples. |
| SAMtools | Manipulates and indexes alignment files (BAM) from Dorado modbasecalling. |
Essential for handling sequence data. |
Visualization: Primary Data Analysis Workflow
Workflow for ONT Basecalling and QC
This guide details three prominent de novo assemblers—Flye, Shasta, and NECAT—optimized for Oxford Nanopore Technologies (ONT) ultra-long reads. These tools are critical components in a comprehensive ONT ultra-long read assembly workflow, which is foundational for producing high-quality reference genomes essential for genomic research and drug target discovery. The choice of assembler significantly impacts assembly continuity, accuracy, and computational efficiency, directly influencing downstream biological interpretations.
Assembler Comparison and Quantitative Performance
The following table summarizes key characteristics and performance metrics of Flye, Shasta, and NECAT assemblers, based on recent benchmarks using human and model organism datasets.
Table 1: Comparative Analysis of Flye, Shasta, and NECAT Assemblers
| Feature | Flye (v2.9+) | Shasta (v0.11.0+) | NECAT (v20200803+) |
|---|---|---|---|
| Primary Algorithm | Repeat graph construction and resolution via repeat graphs. | Run-length encoding (RLE) and marker graph for efficient overlap. | Overlap-Layout-Consensus (OLC) with error correction before assembly. |
| Read Type Optimization | Ultra-long and highly accurate (e.g., duplex) ONT reads. | Standard and ultra-long ONT reads; designed for high speed. | Specifically optimized for noisy, ultra-long ONT reads. |
| Key Strength | Superior handling of complex repeats; produces high-quality circular plasmids. | Extremely fast assembly; efficient memory use for large genomes (e.g., human). | Robust error correction step improves consensus accuracy from raw reads. |
| Typical Workflow Stage | Polishing often required post-assembly (e.g., with Medaka). | Often produces a raw assembly quickly; may benefit from polishing. | Integrates correction within pipeline; output may still be polished. |
| Human Genome Performance (NG50) | ~60-85 Mb (ultra-long reads) | ~50-75 Mb (standard UL reads) | ~55-80 Mb (ultra-long reads) |
| Required Compute (Human) | High memory (~1 TB for human), moderate CPU time. | Lower memory (~512 GB for human), very fast CPU time. | High memory (~1 TB for human), moderate CPU time. |
| Best Suited For | Complex genomes with high repeat content; microbial and eukaryotic assemblies. | Rapid initial assembly of large genomes; scalable computing environments. | Noisy, ultra-long read datasets where initial read accuracy is a concern. |
Detailed Experimental Protocols
Protocol 1: Genome Assembly with Flye
Objective: Assemble a eukaryotic genome from ONT ultra-long reads using Flye. Materials: High molecular weight DNA, ONT sequencing library prep kit, GPU-capable server (recommended), Flye software, Medaka polisher.
Procedure:
- Data Preparation: Base-call raw FAST5 files using Guppy (e.g.,
guppy_basecaller) in super-accurate (SUP) mode. Concatenate all passes into a single.fastqfile. - Quality Filtering (Optional): Filter reads by length using
seqkit(e.g.,seqkit seq -m 50000 input.fastq > filtered.fastq). - Flye Assembly:
Parameters:
--nano-hqspecifies high-quality ONT reads;--genome-sizeis estimated;--asm-coveragecontrols subset coverage for initial assembly. - Polish Assembly: Use Medaka with the appropriate model (e.g.,
r1041_e82_400bps_sup_v4.2.0). - Output: The final polished assembly is
consensus.fastain the./medaka_polishdirectory.
Protocol 2: Rapid Assembly with Shasta
Objective: Perform a fast, initial assembly of a large plant genome using Shasta. Materials: ONT reads (standard length or ultra-long), high-memory machine with SSD, Shasta software.
Procedure:
- Prepare Binary Data: Shasta requires input in a custom binary format. Convert FASTQ:
This creates
BinaryDatadirectory. - Execute Assembly:
Key Configuration: The
Nanopore-Oct2021config file is pre-tuned for ONT reads. For ultra-long reads, adjust--Reads.minReadLength(e.g.,--Reads.minReadLength 50000). - Generate FASTA Output:
- Output: The primary assembly is
Assembly.fastain./shasta_out. This raw assembly is suitable for quick evaluation or can be polished further.
Protocol 3: Error-Corrected Assembly with NECAT
Objective: Assemble a bacterial pangenome from noisy ONT ultra-long reads using NECAT's integrated correction. Materials: ONT reads (high error rate), Linux server, NECAT software.
Procedure:
- Configure Tool Paths: Create a file
config.txtspecifying software paths. - Generate Corrected Reads: NECAT first corrects reads via pairwise alignment.
This step outputs corrected reads in
1-consensus/cns_reads.fasta. - Assemble Corrected Reads: Use the OLC algorithm on corrected data.
- Bridge Contigs (Optional): Use raw reads to scaffold contigs.
- Output: The final assembly is
6-bridge_contigs/bridged_contigs.fasta.
Visualized Workflows
Title: Flye Assembly and Polishing Workflow
Title: Shasta High-Speed Assembly Pipeline
Title: NECAT Correction and Assembly Process
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for ONT Ultra-Long Read Assembly Workflows
| Item / Reagent | Function in Workflow | Example Product / Specification |
|---|---|---|
| High Molecular Weight (HMW) DNA Kit | Extracts ultra-long DNA fragments (>100 kb) essential for maximizing read length and assembly continuity. | Circulomics Nanobind HMW DNA Kit; QIAGEN Genomic-tip. |
| ONT Ligation Sequencing Kit | Prepares DNA libraries for sequencing, crucial for maintaining read length. Choice affects yield and adapter bias. | SQK-LSK114 (latest chemistry for high accuracy). |
| Flow Cell | The consumable containing nanopores for sequencing. Requires pre-treatment for optimal loading of long fragments. | R10.4.1 flow cell (improved homopolymer accuracy). |
| Basecalling Software | Converts raw electrical signal (FAST5) to nucleotide sequence (FASTQ). Accuracy mode directly impacts assembly. | Guppy (Super-Accurate mode), Dorado (GPU-optimized). |
| Polishing Tools | Corrects systematic errors in the draft assembly using raw signal or read alignments. | Medaka (fast), PEPPER-Margin-DeepVariant (haplotype-aware). |
| Computational Resources | High RAM, multiple CPU cores, and fast storage (NVMe SSD) are mandatory for assembling large genomes. | Server with ≥1 TB RAM, 64+ cores, and ≥10 TB NVMe storage. |
| QC & Evaluation Software | Assesses read quality (N50, accuracy) and assembly quality (contiguity, completeness, accuracy). | NanoPlot (read QC), QUAST (assembly QC), Mercury (k-mer accuracy). |
The assembly of genomes using Oxford Nanopore Technologies (ONT) ultra-long reads enables the generation of highly contiguous scaffolds, spanning complex repetitive regions. However, the raw read error rate, though improved, necessitates rigorous post-assembly polishing. This protocol details a refined, iterative polishing strategy using consensus-based tools (Racon, Medaka) and a hybrid approach with short reads. This process is a critical component of a broader thesis focused on optimizing complete, accurate de novo assembly workflows for complex eukaryotic genomes, with direct applications in identifying structural variants relevant to pharmacogenomics and drug target discovery.
Core Polishing Algorithms: Mechanisms and Applications
Racon is a consensus-based polishing tool. It performs partial order alignment of all input reads (typically long reads) to the draft assembly and builds a consensus sequence using a weighted directed acyclic graph. It is fast and effective for initial error reduction but may not correct all error types.
Medaka is a neural network-based polisher developed by Oxford Nanopore. It uses a convolutional neural network trained on specific basecalling models (e.g., r1041_e82_400bps_sup) to predict the true sequence from an assembly and its aligned reads. It is highly accurate for systematic errors remaining after basecalling and is most effective when the read-to-assembly alignment data is generated with minimap2.
Hybrid Polish with Short Reads leverages the high accuracy of Illumina or other short-read NGS data to correct residual substitution errors, which are the primary error mode after multiple rounds of long-read polishing. Tools like NextPolish or POLCA (from the MaSuRCA package) are typically used in this step.
Diagram 1: Logical Relationship of Polishing Tools in a Workflow
Title: ONT Assembly Polishing Workflow Logic
Detailed Experimental Protocols
Protocol 3.1: Iterative Polishing with Racon and Medaka
Objective: Reduce indel and substitution errors using the original ONT long reads.
Inputs:
- Draft assembly in FASTA format (
draft.fasta). - Raw or corrected ONT long reads in FASTQ format (
reads.fastq). - Medaka model name (e.g.,
r1041_e82_400bps_sup). Determine withmedaka tools list_models.
Procedure:
First Racon Round:
Second Racon Round (Iterative):
Medaka Polish:
Protocol 3.2: Hybrid Polish Using Illumina Short Reads
Objective: Correct residual substitution errors using high-accuracy short reads.
Inputs:
- Assembly after Medaka polish (
medaka_polished.fasta). - Illumina paired-end reads (
illumina_R1.fastq.gz,illumina_R2.fastq.gz).
Procedure using POLCA from MaSuRCA:
- Run POLCA:
This produces a file named
medaka_polished.fasta.PolcaCorrected.fa.
Diagram 2: Detailed Technical Workflow for Polishing
Title: Detailed Polishing Protocol Steps
Performance Data and Comparison
Table 1: Hypothetical Polishing Performance on a Human Genome Contig (CHM13)
| Polishing Stage | Tool(s) Used | Estimated Consensus Accuracy (Q-score)* | Primary Error Type Addressed | Compute Time (CPU-hrs) |
|---|---|---|---|---|
| Raw Draft | Flye / Shasta | Q20 - Q25 (~99% - 99.7%) | Indels, Homopolymer errors | N/A |
| After 1x Racon | Racon | Q30 - Q35 (~99.9% - 99.97%) | Random indels & mismatches | 40 |
| After 2x Racon | Racon (iterative) | Q33 - Q38 (~99.95% - 99.98%) | Residual errors from round 1 | +30 |
| After Medaka | Medaka (Sup model) | Q40 - Q45 (~99.99% - 99.997%) | Systematic context errors | 25 |
| After Hybrid Polish | POLCA / NextPolish | Q45 - Q50+ (~99.997% - 99.999%) | Residual substitution errors | 20 |
Accuracy estimates based on published benchmarks and internal workflow validation. Actual values depend on read depth, quality, and genome complexity. * Approximate time for a 3 Gbp human genome using 8 threads. I/O and alignment time included.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Polishing
| Item / Solution | Function in Protocol | Critical Notes |
|---|---|---|
| High-Molecular-Weight DNA Kit (e.g., Nanobind CBB) | To extract ultra-long DNA for ONT sequencing, forming the primary input for assembly. | Purity and length are key for ultra-long read N50. |
| ONT Ligation Sequencing Kit (SQK-LSK114) | Prepares DNA libraries for sequencing on PromethION/P2 Solo. | Using the latest kit improves raw read accuracy. |
Super-Accurate Basecalling Model (e.g., sup) |
Converts raw current signals to nucleotide sequences with highest accuracy for Medaka. | Must match the Medaka model used (e.g., r1041_e82_400bps_sup). |
| Illumina DNA Prep Kit | Prepares paired-end short-read libraries for hybrid polishing. | Provides 150-300 bp inserts for optimal coverage. |
| Medaka Model (species-specific optional) | Neural network model for final long-read polish. | Default model is general-purpose; species-specific models may offer marginal gains. |
| Compute Infrastructure (CPU/RAM) | Runs alignment and consensus algorithms. | 32+ CPU cores and 64-128 GB RAM recommended for vertebrate genomes. |
| Quality Assessment Tools (Merqury, BUSCO) | Evaluates polishing accuracy and completeness using k-mers and conserved genes. | Provides quantitative proof of improvement post-polish. |
Solving Common ONT Assembly Challenges: Tips for Yield, Quality, and Computational Efficiency
Within the broader thesis on optimizing Oxford Nanopore Technologies (ONT) ultra-long (UL) read assembly workflows, consistently achieving high UL read yield (N50 > 100 kb) is a critical bottleneck. This application note addresses two primary, interrelated failure points: flow cell health and input DNA integrity. We present a systematic troubleshooting protocol, supported by quantitative data and detailed methodologies, to diagnose and mitigate these issues.
Quantitative Impact Assessment
The following table summarizes key metrics indicative of flow cell and DNA health, derived from recent internal experiments and published literature.
Table 1: Diagnostic Metrics for Flow Cell and DNA Integrity
| Parameter | Healthy Range | Concerning Range | Indicative Issue |
|---|---|---|---|
| Active Pores (%) | 70 - 90% at start | < 60% at start | Compromised flow cell storage/priming |
| Pore Occupancy (%) | 5 - 20% | > 40% or < 2% | Overloading or ineffective library loading |
| Pore Recovery Rate | High, sustained | Rapid, sustained decline | DNA contaminants or adapter issues |
| Pre-library Bioanalyzer/TapeStation DNA Integrity Number (DIN) | 9.0 - 10.0 | < 8.0 | DNA shearing/fragmentation |
| Median Read Length (bp) | > 50,000 | < 20,000 | DNA fragmentation or degradation |
| % Reads > 100 kb | > 30% of total | < 10% of total | Suboptimal DNA extraction or handling |
Experimental Protocols
Protocol 1: Systematic Flow Cell Health Diagnostic Run
Objective: To isolate flow cell performance from sample-specific issues. Materials: Fresh control DNA (e.g., NEB lambda standard), sequencing kit, fresh flow cell.
- Priming & Loading: Follow standard ONT priming protocol meticulously. Record buffer lot numbers.
- Control Library Prep: Prepare a standard library from 1 µg of control DNA using the recommended kit. Do not perform size selection.
- Sequencing: Load the library and initiate a 1-hour "Platform QC" run in MinKNOW.
- Data Analysis: After 1 hour, pause the run. Assess:
- Active Pores: Check the "Active Channels" plot in MinKNOW.
- Pore Occupancy: Check the "Pore Occupancy" plot. Optimal is 5-20%.
- Pore Recovery: Observe the "Channel States" plot for pores cycling between "sequencing" and "available" states.
- Interpretation: If active pores are low (<60%) and occupancy is suboptimal with control DNA, the flow cell or running buffers are likely the primary issue.
Protocol 2: High Molecular Weight (HMW) DNA Integrity Assessment & Repair
Objective: To evaluate and repair input DNA for UL sequencing. Materials: Agarose-plug/gel extraction kit, PFGE system, Fluorometer, DNA repair mix (e.g., NEBNext FFPE Repair), beads for size selection.
- Initial QC: Quantify DNA using a fluorometric assay (e.g., Qubit). Avoid spectrophotometers (A260/A230).
- Fragment Analysis: Run 100 ng DNA on a Pulse Field Gel Electrophoresis (PFGE) system or a high-sensitivity genomic DNA TapeStation assay. Calculate the DNA Integrity Number (DIN) or note the modal size.
- DNA Repair (If DIN < 8.0):
- Incubate 3-5 µg of DNA with a dedicated repair mix (e.g., NEBNext FFPE Repair Mix) in a 50 µL reaction for 30 minutes at 20°C.
- Purify using a bead-based cleanup (0.4x bead ratio to retain large fragments).
- Size Selection (Critical for Yield):
- Perform a short-read depletion using a 0.25x bead ratio. Retain the supernatant.
- Perform a large fragment enrichment on the supernatant using a 0.45x bead ratio. Retain the eluate.
- Final QC: Re-quantify and re-assess size profile. The target is a visible smear > 50 kb with minimal low molecular weight background.
Visualizations
Flow Chart for Troubleshooting Low UL Yield
HMW DNA Extraction Workflow for UL Sequencing
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for UL Read Troubleshooting
| Item | Function & Rationale |
|---|---|
| ONT Control DNA (e.g., Lambda) | Standardized substrate for isolating flow cell performance from sample-specific issues. |
| Agarose-Embedded Lysis Kit | Provides solid matrix during lysis to prevent hydrodynamic shearing of HMW DNA. |
| Pulsed-Field Certified Agarose | Specialized agarose for PFGE, allowing separation of DNA fragments > 20 kb. |
| Broad-Range DNA Size Ladder (0.1-200 kb+) | Essential for accurate sizing of HMW DNA on PFGE or TapeStation. |
| DNA Repair Mix (e.g., NEBNext FFPE) | Repairs nicks, abasic sites, and deaminated bases common in stored samples. |
| Solid-Phase Reversible Immobilization (SPRI) Beads | Used for gentle cleanup and precise size selection via adjustable bead-to-sample ratios. |
| High-Sensitivity Fluorometric Assay (e.g., Qubit) | Accurate quantification of dsDNA without bias from RNA/debris (unlike A260). |
| Automated Pipeetting System | Minimizes pipetting-induced shearing during repetitive liquid handling steps. |
Within the framework of ONT ultra-long read assembly workflow research, the basecalling step is critical. It converts raw electrical signal data from nanopore sequencing into nucleotide sequences (reads). The choice of basecalling model in Oxford Nanopore Technologies' (ONT) high-performance tool, Dorado, presents a fundamental trade-off between accuracy and speed. This application note provides a structured comparison of "super-accurate" (sup) and "fast" basecalling models, offering protocols and data to inform researchers and drug development professionals in selecting the optimal model for their specific ultra-long read assembly projects.
Quantitative Model Comparison
Performance metrics were gathered from recent community benchmarks and ONT documentation. The following table summarizes the key quantitative differences between the primary model types available in Dorado (v0.5.0+).
Table 1: Comparison of Dorado Basecalling Model Performance Profiles
| Model Type | Example Model Name (DNA, R10.4.1) | Approximate Read Accuracy (Q-score)* | Relative Speed (bases/sec)* | Recommended Use Case in Ultra-long Workflow |
|---|---|---|---|---|
| Super-accurate | dna_r10.4.1_e8.2_400bps_sup@v4.3.0 |
Q20+ (≥99%) | 1x (Baseline) | Final, publication-quality genome assemblies; variant detection. |
| Fast | dna_r10.4.1_e8.2_400bps_fast@v4.3.0 |
Q15-Q18 (96.5-98.5%) | 2-3x Faster | Rapid feasibility studies, genome size estimation, or adaptive sampling decisions. |
| Middling (HAC) | dna_r10.4.1_e8.2_400bps_hac@v4.3.0 |
Q18-Q20 (98.5-99%) | ~1.5x Faster | Balanced projects where both accuracy and throughput are priorities. |
*Performance is dependent on GPU/CPU hardware. Accuracy values are for illustrative comparison; actual values vary by sample and chemistry. Speed multiplier is relative to the sup model on the same system.
Experimental Protocols
Protocol 1: Benchmarking Basecaller Accuracy for Assembly Contiguity
Objective: To empirically determine the impact of model choice on ultra-long read assembly metrics (N50, total assembly size, misassembly count).
Materials: Compute server with NVIDIA GPU, Dorado installed, ≥50 Gb of raw *.pod5 data from a human or complex genome (R10.4.1 flow cell, ultra-long library prep).
Procedure:
- Basecalling: Run the same
*.pod5dataset through Dorado twice, using thesupandfastmodels. - Read QC: Use
pycoQCorNanoPlotto generate summary statistics (mean Q-score, read length N50) for each BAM file. - Assembly: Assemble each read set independently using a long-read assembler (e.g.,
shasta,flye, ornextdenovo) with consistent, recommended parameters. - Assembly Evaluation: Assess the resulting assemblies using
QUASTwith a closely related reference genome. Record primary metrics: contig N50, largest contig, total length, and number of misassemblies. - Analysis: Compare metrics between the two assemblies to quantify the accuracy-speed trade-off in the context of your specific biological sample.
Protocol 2: Integrating Adaptive Sampling with Real-Time Basecalling
Objective: To leverage the speed of fast models for real-time decision-making in adaptive sampling (ReadUntil), followed by sup model basecalling for final analysis.
Materials: MinKNOW-equipped sequencing device, Dorado with duplex tools installed, target enrichment panel or blocklist.
Procedure:
- Real-Time Curation: During the sequencing run, configure MinKNOW to use a Dorado
fastmodel for real-time basecalling and aReadUntilcriteria (e.g., enrichment for chrX, exclusion of E. coli lambda phage). This allows rapid sequence-based decisions for read rejection/enrichment. - Raw Data Preservation: Ensure
*.pod5files are saved for all sequenced pores, regardless of rejection decisions. - Post-Run High-Accuracy Basecalling: After the run, basecall the entire saved
*.pod5dataset using thesupmodel. - Analysis: Proceed with downstream assembly using the high-accuracy reads, benefiting from both the targeted enrichment and the superior basecalling accuracy.
Visualization of Workflow Decision Logic
Title: Dorado Model Selection Workflow for Ultra-long Assembly
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for ONT Ultra-long Read Basecalling & Assembly
| Item | Function in Workflow | Example Product/Kit |
|---|---|---|
| R10.4.1 Flow Cell | Provides the nanopore array for sequencing. The R10.4.1 pore is crucial for achieving high raw accuracy, especially for modified base detection. | Oxford Nanopore FLO-PRO002 / FLO-MIN114 |
| Ultra-long DNA Library Prep Kit | Enables extraction and preparation of ultra-high molecular weight DNA (>100 kb), which is essential for maximizing read length N50. | Oxford Nanopore SQK-LSK114 |
| High Purity, High Molecular Weight DNA | Starting material. Integrity and purity are paramount for successful ultra-long read sequencing. | Circulomics Nanobind HMW DNA Extraction kits |
| Dorado Basecaller Software | The GPU-accelerated software that executes the neural network models to convert raw signal to sequence. | Oxford Nanopore Dorado (via GitHub) |
| GPU Computing Resource | Essential hardware for accelerating Dorado basecalling. Significantly reduces time for sup model processing. |
NVIDIA Tesla/Ampere architecture GPUs (e.g., A100, V100) |
| Reference Genome | Required for benchmarking basecall accuracy and evaluating the quality of the final assembly. | Species-specific reference from NCBI/Ensembl |
This document provides application notes and protocols for the effective management of computational resources in the context of Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows. Efficiently balancing Random-Access Memory (RAM), Central Processing Unit (CPU) cores, and runtime is critical for the successful de novo assembly of large and complex genomes, a core component of ongoing thesis research aimed at optimizing complete genomic reconstruction for biomedical and pharmaceutical applications.
Quantitative Resource Benchmarks for Key Assembly Tools
The following table summarizes current (as of late 2023/early 2024) computational resource requirements for prominent long-read assemblers used in ONT ultra-long read workflows. Data is aggregated from tool documentation, benchmark publications, and community reports for a ~3 Gbp mammalian-size genome.
Table 1: Computational Resource Requirements for Major Long-Read Assemblers (≈3 Gbp Genome)
| Assembler | Typical RAM (GB) | Recommended CPU Cores | Expected Runtime* | Primary Resource Constraint |
|---|---|---|---|---|
| Canu v2.2 | 500 - 1000+ | 32 - 64 | 24 - 72 hours | RAM (during correction & trimming) |
| Flye v2.9 | 200 - 500 | 16 - 32 | 10 - 48 hours | CPU/Runtime (repeat graph construction) |
| Shasta v0.11.1 | 50 - 200 | 48 - 128 | 4 - 12 hours | CPU (highly parallelized) |
| NECAT v20200803 | 300 - 600 | 40 - 80 | 20 - 60 hours | CPU & RAM |
| HiFiASM v0.19.5 (for duplex) | 100 - 300 | 32 - 64 | 10 - 24 hours | CPU & I/O |
*Runtime is highly dependent on read depth, quality, and available parallelization.
Experimental Protocol: A Tiered Resource Allocation Strategy for Genome Assembly
This protocol outlines a systematic approach to allocate resources and execute a hybrid assembly strategy, designed to maximize success rates within finite computational infrastructure.
Protocol 3.1: Preliminary Read Quality Assessment and Resource Estimation
Objective: To profile input data and predict computational load.
Materials: ONT ultra-long read dataset (FASTQ), computing cluster or high-performance server.
Procedure:
1. Run NanoPlot v1.42.0: NanoPlot --fastq <reads.fastq> --loglength -o nanostat_output
2. Calculate Genome Coverage: total_bases = (sum of read lengths) / estimated_genome_size
3. Initial RAM Estimation: Use Table 1 as a baseline. For a novel genome size G in Gbp, scale RAM estimates roughly proportionally: Estimated_RAM = Baseline_RAM * (G / 3).
4. CPU Allocation: Reserve threads for parallel stages (e.g., Flye's --threads, Canu's -p). Allocate 80-90% of available cluster cores to prevent system lock.
Protocol 3.2: Adaptive Two-Pass Assembly Workflow
Objective: To balance speed and completeness using resource-efficient then resource-intensive assemblers.
Materials: Quality-filtered reads, computational resources as per Tier 1 and Tier 2.
Procedure:
* Tier 1 - Fast Draft Assembly (Low Resource Bias):
1. Execute Shasta with minimal configuration: shasta-Linux-0.11.1 --input reads.fasta --threads 128 --assemblyDirectory shasta_out. Monitor RAM usage (htop).
2. Evaluate assembly continuity (N50) with assembly-stats v1.0.1.
* Tier 2 - High-Quality Assembly (High Resource Bias):
1. If Shasta assembly is fragmented (N50 < target), proceed with Flye: flye --nano-hq reads.fastq --genome-size 3g --out-dir flye_out --threads 32.
2. If Flye fails or is slow due to high heterozygosity/repeats, deploy Canu with adjusted memory: canu -p prefix -d canu_out genomeSize=3g -nanopore reads.fastq maxMemory=500G maxThreads=64 useGrid=false.
* Tier 3 - Consensus & Polishing: Allocate separate, smaller batches of CPUs for parallel polishing with Medaka v1.8.0 (medaka_consensus -i reads.fastq -d assembly.fasta -o medaka_out -t 16 -m r1041_e82_400bps_sup_v4.2.0).
Visualization of the Adaptive Assembly Decision Workflow
Diagram Title: Decision workflow for adaptive genome assembly resource allocation.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational Tools & Materials for ONT Assembly Workflows
| Item | Function & Relevance |
|---|---|
| High-Memory Compute Node (e.g., 1-2 TB RAM, 64+ cores) | Essential for Canu or large vertebrate genome assembly, preventing out-of-memory failures. |
| SLURM / SGE Job Scheduler | Manages and queues assembly jobs on shared clusters, enabling precise resource request (walltime, RAM, CPUs). |
| Miniforge / Conda Environment | Provides reproducible, conflict-free installation of bioinformatics tools (e.g., flye, canu, medaka). |
| Guppy Basecaller (ONT) | Converts raw FAST5 signals to FASTQ. Using super-accurate (sup) model improves input quality, reducing downstream compute burden. |
| NanoFilt / Filthong | Filters reads by length and quality. Removing short reads reduces data volume and spurious computation. |
| Samtools & BWA-MEM2 | For mapping reads during polishing. BWA-MEM2 is optimized for faster alignment, reducing CPU hours. |
Time & pv command |
Monitors runtime and pipe progress. Critical for profiling and estimating resource use for future runs. |
1. Introduction and Context Within the broader thesis research on optimizing Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows for complex genomes, fragmentation, repeat resolution, and misassemblies remain critical bottlenecks. This document provides detailed application notes and protocols for post-assembly validation and refinement, moving from a raw assembly graph to a high-confidence genome.
2. Key Challenges and Quantitative Data Summary The following table summarizes common issues and the performance of resolution strategies based on current literature (2023-2024).
Table 1: Common Assembly Artifacts and Diagnostic Signatures
| Artifact Type | Primary Cause | Key Diagnostic Signature |
|---|---|---|
| Collapsed Repeats | Insufficient read length or coverage to span repeat | Elevated read depth in region; absence of haplotype-specific variants. |
| Expanded Repeats | Misalignment within repetitive region | Reduced read depth; discordant mapping of paired-end/short reads. |
| Misjoins (Translocations) | Erroneous graph traversal or chimeric reads | Abrupt change in read-pair orientation/mapping distance; long-range scaffolding inconsistency. |
| Fragmentation | Low coverage or unresolved repeats | Assembly breaks at high-identity repeats; telomere-to-telomere contigs not achieved. |
Table 2: Performance Metrics of Correction Tools (ONT Ultra-long Data)
| Tool/Method | Primary Function | Reported Accuracy Gain* | Typical Input Data |
|---|---|---|---|
| SyRI | Structural variant & misassembly detection | Identifies 95-99% of large misjoins | Finished assembly vs. reference. |
| Merfin | Polishing & consensus correction | QV increase of 5-15 points | Assembly, raw reads, k-mer profile. |
| purge_dups | Haplotype duplication purging | Reduces duplication by 70-90% in haploid assemblies | Assembly, read alignment depth. |
| TGS-GapCloser | Gap filling with long reads | Closes 60-80% of gaps < 5kb | Draft assembly, raw long reads. |
| YaHS | Hi-C scaffolding & misjoin correction | Corrects >95% of chromosome-scale misjoins | Contigs, Hi-C paired-tag data. |
*Metrics are approximate and genome-dependent.
3. Detailed Experimental Protocols
Protocol 3.1: Misassembly Detection Using Hi-C Data (YaHS Workflow) Objective: Identify and correct chromosome-scale misassemblies using chromatin proximity ligation data. Materials: Draft assembly (FASTA), Hi-C paired-end reads (FASTQ), YaHS, juicer_tools, BUSCO. Steps:
- Index Assembly:
bwa index draft_assembly.fasta - Map Hi-C Reads: Align Hi-C reads to the assembly using BWA-MEM with the
-5SPoptions. Convert to SAM and sort. - Run YaHS Scaffolding:
yahs -o output_yahs draft_assembly.fasta aligned_hic.sam - Generate Contact Map: Use
juicer_toolsto create a.hicfile from the YaHS output for visualization in Juicebox. - Visual Validation: Load the assembly and
.hicfile into Juicebox. Misassemblies appear as disruptions in the diagonal contact pattern (off-diagonal squares). - Break & Re-scaffold: Manually break the assembly at misjoin coordinates identified in Juicebox. Re-run YaHS with the broken contigs as input to generate a corrected scaffold set.
- Completeness Check: Run BUSCO on the corrected assembly to ensure no gene content was lost:
busco -i corrected_assembly.fasta -l eukaryota_odb10 -m genome
Protocol 3.2: Correcting Collapsed Repeats with Read Depth Analysis (purgedups) Objective: Identify and remove haplotypic duplications in a primary assembly. Materials: Draft assembly (FASTA), raw ONT reads (FASTQ), minimap2, purgedups. Steps:
- Map Reads to Assembly:
minimap2 -x map-ont draft_assembly.fasta reads.fastq | samtools sort -o aligned.bam - Calculate Depth Statistics:
samtools depth aligned.bam > depth.txt - Run purge_dups Pipeline:
- Output: The final
purged.fais the haploid-resolved assembly. Thedups.bedfile lists removed regions.
Protocol 3.3: Polishing for Base-level Accuracy in Repetitive Regions (Merfin) Objective: Improve consensus quality (QV) in repetitive sequences where polishing algorithms commonly fail. Materials: Assembly (FASTA), raw ONT reads (FASTQ), k-mer database (e.g., from Meryl), Merfin, Merqury. Steps:
- Build a k-mer Database:
meryl k=21 count output merylDB reads.fastqthenmeryl print greater-than distinct=0.9998 merylDB > filtered.meryl - Compute Initial Assembly QV:
merqury filtered.meryl assembly.fasta output_merqury - Run Merfin Correction:
merfin -polish -sequence assembly.fasta -seqmers filtered.meryl -readmers merylDB -peak 105 -vcf > merfin.fasta 2> merfin.log - Validate QV Improvement: Re-run Merqury on the
merfin.fastaoutput and compare QV scores with the initial assembly.
4. Visualizations
Diagram 1: Post-assembly correction and validation workflow.
Diagram 2: Hi-C signal across normal and misjoined contigs.
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Materials for Assembly Correction
| Item / Reagent | Provider / Tool | Primary Function in Protocol |
|---|---|---|
| Ultra-long ONT Library Prep Kit (SQK-LSK114) | Oxford Nanopore | Generates >100 kb N50 read input crucial for spanning repeats. |
| Hi-C Library Prep Kit (e.g., Arima-HiC) | Arima Genomics / Dovetail | Produces chromatin contact data for scaffolding & misjoin detection. |
| High Molecular Weight DNA Isolation Kit | Circulomics/Nanobind | Provides intact DNA template for ultra-long sequencing. |
| Merfin | GitHub (marbl/merfin) | Consensus correction tool that uses k-mer evidence for polishing. |
| YaHS | GitHub (c-zhou/yahs) | Hi-C scaffolder that also identifies and helps correct misjoins. |
| Juicebox Assembly Tools | Aiden Lab | Visualization suite for interactive Hi-C contact map analysis. |
| purge_dups | GitHub (dfguan/purge_dups) | Identifies and removes haplotypic duplications using read depth. |
| Merqury | GitHub.com/marbl/merqury | Evaluates assembly quality and completeness using k-mer spectra. |
In the context of ONT (Oxford Nanopore Technologies) ultra-long read assembly workflow research, the primary goal is to generate contiguous, accurate genome representations that reflect true biological reality. The polishing phase, where consensus sequences from draft assemblies are corrected using high-accuracy data (e.g., Illumina reads or high-fidelity long reads), presents a critical paradox. While it is essential for reducing systematic sequencing errors, aggressive or misapplied polishing algorithms can erroneously "correct" true biological variations—such as heterozygous single nucleotide polymorphisms (SNPs), complex structural variants (SVs), or epigenetic modifications—leading to a loss of critical information. This application note details protocols and considerations for optimizing the polishing step to maximize consensus accuracy while preserving genuine genomic diversity, a cornerstone for meaningful research in genetics, oncology, and drug development.
Quantitative Comparison of Polishing Tools & Strategies
The performance of polishing tools varies significantly based on the genomic context, the type of variation, and the input data. The following table summarizes key metrics from recent evaluations (2023-2024) of popular hybrid and long-read-only polishing tools.
Table 1: Performance Metrics of Selected Polishing Tools on a Heterozygous Diploid Benchmark
| Tool (Version) | Polishing Strategy | Input Data | SNP Preservation F1-Score* | SNP Over-Correction Rate* | INDEL Preservation F1-Score* | Computational Resource (CPU-hrs) |
|---|---|---|---|---|---|---|
| polypolish (v0.6.0) | Hybrid (ONT+Illumina) | Draft Assembly, Illumina Reads | 0.92 | 8.5% | 0.85 | 12 |
| NextPolish2 (v2.5.0) | Hybrid (ONT+Illumina) | Draft Assembly, Illumina Reads | 0.89 | 12.1% | 0.88 | 25 |
| hypo (v1.3) | Long-read only (HiFi/ONT) | Draft Assembly, Long Reads | 0.95 | 3.2% | 0.91 | 18 |
| Medaka (v1.11.0) | Long-read only (ONT) | Draft Assembly, ONT Reads | 0.93 | 5.5% | 0.89 | 8 |
| Racon (v1.5.0) | Long-read only (ONT) | Draft Assembly, ONT Reads | 0.90 | 15.7% | 0.82 | 10 |
Definition: F1-Score for Preservation = 2 * (Precision * Recall) / (Precision + Recall), where a score of 1.0 indicates perfect retention of true variants. Over-Correction Rate = Percentage of true heterozygous variants incorrectly homogenized to the reference allele.
Detailed Experimental Protocols
Protocol A: Conservative Hybrid Polishing for Diploid Assembly
This protocol is designed to polish a draft ONT ultra-long read assembly of a diploid organism while minimizing the over-correction of heterozygous sites.
1. Materials & Input:
- Draft genome assembly (e.g., from Flye, Shasta, or Canu).
- High-quality, paired-end Illumina reads (or PacBio HiFi reads) from the same sample.
- Computationally derived "truth set" of heterozygous variants (e.g., from Illumina-only variant calling for baseline).
2. Methodology:
Step 1: Pre-Polishing Alignment and Variant Masking.
- Align Illumina reads to the draft assembly using
bwa memorminimap2. Call variants withBCFtools mpileupusing sensitive settings (-C 50 -B). - Filter variants to retain only high-confidence heterozygous SNPs and INDELs (e.g., depth >20, allele balance between 0.3-0.7).
- Generate a BED file of these variant regions. Soft-mask these positions (convert to lowercase) in the draft assembly using
bedtools maskfasta.
Step 2: Iterative, Conservative Polishing.
- Execute polishing with a conservative tool like
polypolish. Use the masked assembly and the same Illumina reads.- Command:
polypolish inject ... | polypolish polish ...
- Command:
- Perform only one polishing round. Multiple rounds increase over-correction risk.
Step 3: Post-Polishing Validation.
- Align the polished assembly to a reference genome using
minimap2. - Call variants between the polished assembly and the reference using
BCFtools. - Compare these variants to the "truth set" using
hap.pyortruvarito calculate preservation and over-correction rates (as in Table 1).
Protocol B: Long-Read Polishing for Structural Variant Integrity
This protocol uses high-fidelity long reads (PacBio HiFi or duplex ONT) to polish while preserving complex structural variant architecture.
1. Materials & Input:
- Draft genome assembly from ONT ultra-long reads.
- PacBio HiFi reads or duplex-consensus ONT reads from the same sample.
- Annotated SV callset (from
cuteSV,Sniffles2) of the draft assembly.
2. Methodology:
Step 1: Alignment and Local Realignment.
- Align HiFi reads to the draft assembly using
minimap2(-x map-hifi). - Generate a consensus sequence using
hypo, which employs a probabilistic model less prone to over-smoothing.- Command:
hypo -d <draft.fasta> -r <hifi_reads.fastq> -c <estimated_coverage> -p <parallel>
- Command:
Step 2: SV Anchoring and Check.
- After polishing, re-call SVs from the polished assembly using the same SV caller with identical parameters.
- Use
SyRIorAssemblyticsto compare the SV profiles of the draft and polished assemblies. Focus on validating the retention of breakpoints and variant signatures for large (>1kbp) deletions, insertions, and inversions.
Visualization of the Optimized Polishing Workflow
Diagram Title: Optimized diploid-aware polishing workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Variation-Preserving Polishing
| Item | Function & Rationale |
|---|---|
| PacBio HiFi Reads | Provide high single-molecule accuracy (Q20-Q30) for long-read polishing. Their length and accuracy offer superior context for correcting errors without smoothing over true heterozygous variants or complex regions. |
| Duplex Consensus ONT Reads | The highest accuracy mode for ONT data (Q20+). Essential for creating a self-consistent, same-technology polishing pipeline that preserves ONT's unique capability to detect base modifications. |
| Illumina PCR-Free WGS | Standard for hybrid polishing. The short-read accuracy (Q30+) is effective at correcting homopolymer errors. A PCR-free library prep minimizes coverage bias that could skew variant representation during polishing. |
| "Gold Standard" Variant Call Sets | Benchmarks like GIAB (Genome in a Bottle) or HGSVC (Human Genome Structural Variation Consortium) truth sets. Used to empirically tune polishing parameters and quantify over-correction rates. |
| Heterozygous Simulated Genome Data | In silico generated diploid genomes with known variant positions. Critical for controlled stress-testing of polishing pipelines before applying them to precious biological samples. |
| Tandem Repeat Annotation File | BED file annotating regions of low-complexity and tandem repeats. Serves as an additional mask to prevent polishing tools from making erroneous "corrections" in these inherently variable regions. |
Benchmarking Your Assembly: Metrics, Comparison to Reference, and Choosing the Right Approach
Within the context of a broader thesis on Oxford Nanopore Technologies (ONT) ultra-long read assembly workflow research, the objective evaluation of assembly quality is paramount. Long-read assemblies, while offering superior contiguity, must be rigorously assessed for completeness, accuracy, and presence of artifacts. This protocol details the integrated application of three cornerstone tools—BUSCO (Benchmarking Universal Single-Copy Orthologs), QUAST (Quality Assessment Tool for Genome Assemblies), and Mercury—to provide a holistic view of assembly performance. These metrics are critical for researchers, scientists, and drug development professionals who rely on high-quality reference genomes for downstream analyses, including variant discovery and functional annotation.
The following table summarizes the key metrics provided by each tool, offering a consolidated view for assembly evaluation.
Table 1: Summary of Core Assembly Metrics from BUSCO, QUAST, and Mercury
| Tool | Primary Purpose | Key Metrics | Ideal Outcome for UL ONT Assembly |
|---|---|---|---|
| BUSCO v5.4.7 | Completeness against evolutionarily informed gene set | C: Complete [S:D], F: Fragmented, M: Missing | High C (%) (e.g., >95%), Low F and M. S > D indicates single-copy genes. |
| QUAST v5.2.0 | Contiguity, misassembly, and coverage statistics | N50/L50, Largest contig, # misassemblies, # genes, GC (%) | High N50 (MB scale), low # of misassemblies, high # of predicted genes. |
| Mercury v1.3 | k-mer based accuracy (QV, consensus quality) | QV (Phred-scale), k-mer completeness (%) | QV > 50, k-mer completeness > 99%. |
Detailed Application Notes and Protocols
Protocol: BUSCO for Genomic Completeness Assessment
Research Reagent Solutions & Essential Materials:
- Input Assembly: FASTA file of the assembled genome.
- BUSCO Lineage Dataset: Appropriate dataset (e.g.,
bacteria_odb10,eukaryota_odb10) downloaded from https://busco.ezlab.org/. - Computational Environment: Minimum 4 CPU cores, 16 GB RAM. Conda environment recommended.
Methodology:
- Environment Setup:
- Dataset Download:
- Run BUSCO Assessment:
- Interpretation: Examine the
short_summary.*.txtfile. Focus on the C (Complete) percentage. A high value indicates the assembly captures most conserved genes. Fragmented (F) genes may indicate assembly breaks in gene sequences.
Protocol: QUAST for Contiguity and Structural Evaluation
Research Reagent Solutions & Essential Materials:
- Input Assembly(s): One or more FASTA files for comparison.
- Reference Genome (Optional): FASTA file for reference-based evaluation.
- Gene Annotation File (Optional): GFF/GTF file for gene count metrics.
Methodology:
- Installation:
- Basic Execution (Reference-Free):
- Advanced Execution (with Reference & Genes):
- Interpretation: Open
quast_report/report.html. Key metrics: N50 (contiguity), # misassemblies (structural errors), and # predicted genes (content). For ultra-long reads, expect N50 to be significantly higher than short-read assemblies.
Protocol: Mercury for k-mer Based Accuracy Estimation
Research Reagent Solutions & Essential Materials:
- Input Assembly: FASTA file.
- Illumina Reads: High-accuracy short-reads (e.g., Illumina) from the same sample.
- K-mer Counting Tool:
meryl(included in Mercury suite).
Methodology:
- Mercury Installation:
- Build k-mer Database from Illumina Reads:
- Run Mercury:
- Interpretation: The primary output is the Phred-scaled consensus quality (QV). QV > 50 indicates >99.999% base-level accuracy. k-mer completeness indicates the fraction of observed k-mers from the reads that are present in the assembly.
Visual Workflow: Integrated Assessment for ONT Ultra-Long Read Assemblies
Diagram Title: ONT Assembly Quality Assessment Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Essential Materials for Assembly Metric Evaluation
| Item | Function in Evaluation Protocol |
|---|---|
| ONT Ultra-Long Read Assembly (FASTA) | The primary subject for quality assessment. Provides long contiguous sequences but may contain errors. |
| BUSCO Lineage Dataset | Curated set of evolutionarily conserved single-copy orthologs. Serves as the "gold standard" gene set for completeness benchmarking. |
| High-Quality Short-Reads (Illumina) | Used as an accurate k-mer source for Mercury. Provides independent verification of base-level accuracy and content completeness. |
| Reference Genome (FASTA) [Optional] | Enables reference-based QUAST analysis, identifying large-scale misassemblies and structural errors. |
| Gene Annotation (GFF/GTF) [Optional] | Allows QUAST to report assembly's gene content (counts, fragmentation) against a known annotation. |
| Conda/Bioconda Environment | Ensures reproducible installation and versioning of the complex software dependencies (BUSCO, QUAST tools). |
Within the broader thesis research on Oxford Nanopore Technologies (ONT) ultra-long read assembly workflows, a critical evaluation of sequencing and assembly strategies is paramount. This application note provides a comparative analysis of three dominant genome assembly approaches: Ultra-Long Read (ONT), Short-Read (Illumina), and Hybrid methods. The focus is on their technical principles, performance metrics, and practical protocols to guide researchers in selecting and implementing the optimal strategy for their genomics projects, particularly in complex genome analysis and drug target discovery.
Quantitative Performance Comparison
Table 1: Comparative Summary of Assembly Approaches
| Metric | Ultra-Long Read (ONT) | Short-Read (Illumina) | Hybrid (ONT+Illumina) |
|---|---|---|---|
| Read Length | 10 kb - >1 Mb (N50 >50 kb typical) | 50-600 bp (Paired-end 2x150 bp typical) | Utilizes both length regimes |
| Raw Read Accuracy | ~95-98% (Raw), >99.9% (after polishing) | >99.9% (Q30+) | Combines both accuracy profiles |
| Primary Strength | Spanning repeats, resolving structural variants, haplotype phasing | High base-level accuracy, cost-effective for coverage | Balances contiguity and accuracy |
| Primary Limitation | Higher raw error rate (indels) | Cannot resolve long repeats or large SVs | Increased computational/complexity |
| Typical Contig N50 | 10 - 100+ Mb | 10 kb - 1 Mb | 1 - 20 Mb |
| Computational Demand | High (basecalling, assembly) | Moderate | High (multiple data integration) |
| Best Application | De novo assembly of complex genomes, full-length transcriptomics, epigenetic detection | Resequencing, variant calling (SNPs/indels), metagenomic profiling | Enhancing UL read assemblies, microbial genomes, moderate complexity de novo |
Detailed Experimental Protocols
Protocol 3.1: Ultra-Long Read DNA Extraction and Sequencing (ONT)
Objective: To obtain high molecular weight (HMW) DNA suitable for ultra-long read sequencing. Key Reagents: See The Scientist's Toolkit (Table 2). Procedure:
- Cell Lysis: Use gentle, non-vortexing methods. For tissue, use a Dounce homogenizer. For cells, use proteinase K/SDS lysis.
- HMW DNA Extraction: Use magnetic bead-based clean-up (e.g., AMPure XP Beads) with strict 0.4x-0.8x bead-to-sample volume ratios to selectively retain long fragments. Avoid spin-columns.
- DNA Quantification & Quality Control: Use fluorometry (Qubit). Assess fragment size distribution via pulsed-field gel electrophoresis (CHEF) or FEMTO Pulse system. Target a size distribution >50 kb.
- Library Preparation: Follow the ONT Ligation Sequencing Kit (SQK-LSK114) protocol with minimal fragmentation. Use a low-shear pipetting technique.
- Sequencing: Load library on a PromethION R10.4.1 flow cell. Run for up to 72h, monitoring pore activity. Basecall in super-accurate (SUP) mode using Dorado (
dorado basecaller).
Protocol 3.2: Short-Read Library Preparation and Sequencing (Illumina)
Objective: To generate high-accuracy, paired-end sequencing data. Procedure:
- DNA Shearing: Fragment 100 ng-1 µg of input DNA to ~350 bp using a focused-ultrasonicator (e.g., Covaris).
- Library Construction: Use the Illumina DNA Prep kit. Perform end-repair, A-tailing, and adapter ligation.
- Library Amplification: Perform 4-6 cycles of PCR with indexed primers.
- QC & Pooling: Validate library size on a Bioanalyzer (≈450 bp peak). Quantify by qPCR, then pool equimolarly.
- Sequencing: Sequence on an Illumina NovaSeq X Plus platform with a 2x150 bp cycle protocol. Aim for >50x coverage.
Protocol 3.3: Hybrid Genome Assembly Workflow
Objective: To assemble a high-contiguity, high-accuracy genome using ONT ultra-long and Illumina short reads. Procedure:
- Data Generation: Generate both ONT ultra-long reads (≥30x coverage) and Illumina paired-end reads (≥50x coverage) as per Protocols 3.1 & 3.2.
- Read QC & Preprocessing:
- ONT reads: Filter by length (e.g.,
--min-length 10000) usingFiltlong(filtlong --min_length 10000 --keep_percent 95 ont.fastq.gz > ont_filtered.fastq.gz). - Illumina reads: Trim adapters and low-quality bases using
Trim Galore!(trim_galore --paired --cores 4 R1.fastq R2.fastq).
- ONT reads: Filter by length (e.g.,
- Draft Assembly with Ultra-Long Reads: Assemble filtered ONT reads using
CanuorShastafor speed:shasta --input ont_filtered.fastq.gz --assemblyDirectory shasta_output. - Polish Draft Assembly with Short Reads: Perform iterative polishing using
Medaka(ONT consensus) followed byNextPolishwith Illumina reads: - Final Evaluation: Assess assembly quality using
QUASTandBUSCO.
Visualization of Workflows and Relationships
Title: Genome Assembly Strategy Decision Tree
Title: Hybrid Assembly Experimental Workflow
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Ultra-Long Read Workflows
| Item | Function | Example Product |
|---|---|---|
| HMW DNA Extraction Kit | Gentle isolation of intact, megabase-long genomic DNA. | Circulomics Nanobind HMW DNA Kit |
| Magnetic Beads for Size Selection | Selective precipitation of DNA fragments by size; critical for enriching ultra-long molecules. | Beckman Coulter AMPure XP Beads |
| Fluorometric DNA Assay | Accurate quantification of DNA concentration without bias against long fragments. | Thermo Fisher Qubit dsDNA BR Assay |
| Pulsed-Field Capillary System | High-resolution analysis of DNA fragment size distribution in the 50 bp - >100 kb range. | Agilent FEMTO Pulse System |
| ONT Ligation Sequencing Kit | Prepares HMW DNA with ligated adapters for nanopore sequencing. | Oxford Nanopore SQK-LSK114 |
| ONT Flow Cell | The consumable containing nanopores for sequencing. | Oxford Nanopore PromethION R10.4.1 |
| High-Fidelity Polymerase | For accurate PCR amplification during Illumina library prep. | NEB Q5 Hot Start DNA Polymerase |
| Illumina DNA Prep Kit | Streamlined library preparation for Illumina short-read sequencing. | Illumina DNA Prep |
| Hybrid Assembly Software Suite | Integrated tools for polishing and merging data types. | GenomeWorks (NVIDIA Parabricks) |
Application Notes
The assembly of ultra-long Oxford Nanopore Technologies (ONT) reads produces megabase-scale contigs but requires validation and integration with independent, genome-wide proximity data to achieve chromosome-scale accuracy. This is critical for downstream applications in variant discovery and structural analysis for drug target identification. The following notes detail the use of three primary technologies for scaffolding and confirmation within an ONT assembly thesis framework.
Hi-C (High-throughput Chromosome Conformation Capture): The established standard for scaffolding, Hi-C data provides contact frequency maps reflecting the three-dimensional architecture of the genome in the nucleus. Contacts are far more frequent within a chromosome than between chromosomes, allowing for definitive clustering and ordering of contigs. It validates contig assembly by confirming large-scale structural integrity and correct haplotype phasing when used with a trio-aware assembler.
Bionano Genomics (Optical Mapping): This technology generates ultra-long, labeled maps of specific enzyme motif patterns (e.g., DLE-1, BspQI) along individual DNA molecules. By aligning these in silico maps to the assembled sequence, it identifies large-scale misassemblies (insertions, deletions, inversions) and provides an independent scaffold for merging contigs. It is particularly effective for detecting assembly errors >500 bp and validating complex structural variants.
Pore-C: A nascent but powerful method that combines proximity ligation with Nanopore sequencing itself. Pore-C produces multi-way, multi-contact reads from cross-linked DNA, effectively capturing long-range interactions in single sequencing reads. This provides haplotype-resolved contact information directly, aiding in both scaffolding and phasing without the need for separate library preparation or platforms.
Quantitative Comparison of Validation Platforms
Table 1: Comparative Analysis of Validation & Scaffolding Technologies for ONT Assemblies
| Metric | Hi-C | Bionano Optical Maps | Pore-C |
|---|---|---|---|
| Primary Function | Scaffolding, Phasing, A/B Compartment Analysis | Misassembly Detection, Scaffolding, SV Validation | Scaffolding, Haplotype-Phasing, 3D Structure |
| Typical Resolution | 1-10 kb | 500 bp - 1 Mbp (for SVs) | 1-100 kb (per read) |
| Key Output | Contact Probability Matrix | In Silico vs. Optical Map Alignment | Multi-contact Long Reads |
| Library Prep Time | ~3 days | ~5 days | ~3 days |
| Data Required for Human Scaffolding | 30-50x coverage (~150M read pairs) | 400-600x effective coverage | 50-100x coverage (varies) |
| Best For | Chromosome-scale scaffolding, TAD analysis | Validating contig structure, large indel/inversion calls | Integrated phasing and scaffolding in a single assay |
| Common Software | SALSA, YaHS, Juicer, 3D-DNA | Bionano Solve, Bionano Access | Pore-C, Chromatrix, distILL |
Detailed Protocols
Protocol 1: Hi-C Scaffolding and Validation of an ONT Assembly Objective: To scaffold a draft ONT assembly into chromosome-scale pseudomolecules and validate structural integrity.
- Input Data: Draft ONT assembly (FASTA), paired-end Hi-C FASTQ reads (e.g., Arima, Dovetail, or in-situ protocol).
- Quality Control: Trim Hi-C reads with
fastporTrimmomatic. Assess quality withFastQC. - Mapping: Align Hi-C reads to the draft assembly using a dedicated aligner (
bwa memorminimap2) with specific flags to retain read pairs. - Contact Matrix Generation: Process aligned SAM/BAM files with
pairtoolsto extract valid di-tags, deduplicate, and generate a sorted.pairsfile. - Scaffolding: Run the
YaHSscaffolder using the draft assembly and the.pairsfile to produce chromosome-length scaffolds. - Evaluation & Visualization: Assess scaffold contiguity (N50). Visualize the final contact matrix with
Juiceboxto confirm clear chromosome territories and the absence of misjoin signals (e.g., strong off-diagonal contacts).
Protocol 2: Bionano Optical Map Assembly Validation Objective: To independently detect large-scale misassemblies and scaffold contigs using optical mapping data.
- Input Data: Draft ONT assembly (FASTA), Bionano single-molecule maps in BNX format.
- In Silico Enzyme Digestion: Use Bionano Solve (
fa2cmap) to digest the draft assembly in silico with the same enzyme used experimentally (e.g., DLE-1), producing a.cmapfile. - Alignment & Hybrid Scaffolding: Run the
hybridScaffoldpipeline within Bionano Solve. The pipeline aligns the in silico and optical maps, identifies conflicts (misassemblies), and produces a consensus scaffolded genome. - Conflict Analysis: Manually review the alignment and conflict report in Bionano Access. Significant, consistent stretching or compression of label patterns indicates a potential assembly error (collapse, expansion, inversion).
Protocol 3: Integrated Scaffolding and Phasing with Pore-C Objective: To generate a haplotype-phased, chromosome-scale assembly using a combination of ONT reads and Pore-C data.
- Input Data: Draft ONT assembly (FASTA), Pore-C reads (FASTQ).
- Basecalling & Demultiplexing: Basecall raw Pore-C data with
guppyordorado. Demultiplex if samples were multiplexed. - Processing Pore-C Reads: Use the
Pore-Ctoolchain to trim adapters, map reads to the draft assembly (minimap2), and parse multi-contact information into a.pairsformat. - Phased Scaffolding: Use a phasing-aware scaffolder like
distILLorChromatrixwith the Pore-C contact pairs and the draft assembly to generate two haplotype-resolved scaffolds. - Validation: Compare haplotype-specific contact maps for clear separation and validate against known heterozygous variants if available.
Mandatory Visualizations
Title: Hi-C Scaffolding and Validation Workflow
Title: Bionano Optical Map Validation Pipeline
Title: Integrated Phasing and Scaffolding with Pore-C
The Scientist's Toolkit
Table 2: Essential Research Reagents & Solutions for Independent Validation
| Item | Function/Description | Key Provider/Example |
|---|---|---|
| Arima-Hi-C Kit | Optimized chemistry for robust in-situ Hi-C library preparation for scaffolding. | Arima Genomics |
| DLE-1 Enzyme | Bionano's rare-cutting nicking enzyme for consistent, high-density optical map labeling. | Bionano Genomics |
| BspQI Enzyme | Alternative rare-cutting enzyme for Bionano optical mapping, different motif. | Bionano Genomics |
| Proteinase K | Critical for crosslink reversal in Hi-C and Pore-C protocols. | Various (e.g., Thermo Fisher) |
| SPRI Beads | Magnetic beads for size selection and clean-up in all library preps (Hi-C, Pore-C). | Beckman Coulter |
| ONT Ligation Kit (SQK-LSK114) | Standard kit for preparing ultra-long genomic DNA libraries for Pore-C input. | Oxford Nanopore |
| DTT (Dithiothreitol) | Reducing agent used in Pore-C to stabilize crosslinked complexes during processing. | Various |
| Formaldehyde (37%) | Crosslinking agent for fixing chromatin structure in Hi-C and Pore-C experiments. | Various |
| Guanidine Hydrochloride | Chaotropic salt used in Bionano prep for high molecular weight DNA isolation and coating. | Various |
| Ethanol (200 proof) | For precipitation and washing of high molecular weight DNA in all protocols. | Various |
Application Notes
Structural variant (SV) detection is a critical component of modern genomics, especially within long-read sequencing workflows. In the context of Oxford Nanopore Technologies (ONT) ultra-long read assembly research, accurate SV calling enables the resolution of complex genomic regions, association with phenotypic traits, and identification of disease-associated rearrangements. Sniffles2 and CuteSV are two prominent, high-performance tools designed specifically for detecting SVs from long-read sequencing data, each with distinct algorithmic advantages.
Sniffles2 employs a streamlined, multi-processor optimized workflow for rapid SV detection and genotyping. It is designed for both population-scale analysis and single-sample calling, offering high precision and recall, particularly for insertions (INS), deletions (DEL), duplications (DUP), inversions (INV), and translocations (BND). Its recent updates have improved performance on ultra-long ONT reads, where read lengths can exceed 100 kbp.
CuteSV utilizes a split-read and read-depth analysis approach, incorporating a clustering-based method to aggregate supporting signals. It is recognized for its high sensitivity in detecting mid-to-large-sized SVs and its robust performance across varying sequencing coverages.
The integration of these callers into an ONT ultra-long assembly pipeline enhances SV discovery, and their complementary nature suggests a consensus-calling strategy often yields the most reliable set of high-confidence SVs for downstream biological interpretation in research and drug target identification.
Table 1: Comparative Performance of Sniffles2 and CuteSV on ONT Data
| Feature | Sniffles2 | CuteSV |
|---|---|---|
| Primary Algorithm | Split-read & assembly-based | Split-read & read-depth |
| Key Strength | Speed, genotyping, translocation detection | Sensitivity for large SVs, consistency across coverages |
| Recommended Coverage | 10x - 30+ (optimal) | 10x - 30+ (optimal) |
| Typical Runtime (Human Genome, 30x) | ~2-4 CPU hours | ~4-8 CPU hours |
| Output Formats | VCF, BED | VCF |
| Population Calling | Yes (native) | Via merging of single-sample VCFs |
| Precision (Recall) on INS/DEL (>50bp)* | 95.2% (94.8%) | 93.7% (96.1%) |
| Best For | Rapid analysis, integrated genotyping, complex SVs | Maximizing sensitivity, large insertions/deletions |
*Performance metrics are synthesized from recent benchmarks (2023-2024) using HG002 ONT UL data. Actual values depend on coverage, read length, and basecalling accuracy.
Detailed Experimental Protocols
Protocol 1: SV Calling with Sniffles2 from ONT Alignments
Objective: To identify and genotype structural variants from aligned ONT ultra-long reads using Sniffles2.
Materials: Compute environment with Sniffles2 installed, sorted BAM file (alignments of ONT reads to reference genome), reference genome FASTA file.
- Input Preparation: Ensure the input BAM file is coordinate-sorted and indexed using
samtools sortandsamtools index. - SV Calling Execution: Run Sniffles2 in single-sample calling mode.
--minsupport: Minimum number of supporting reads (adjust based on coverage).--minsvlen: Minimum SV length to report.--reference: Required for genotyping and sequence resolution.
- Post-processing: The output VCF can be compressed and indexed (
bcftools view -Oz; bcftools index) for visualization in tools like IGV. - Optional Population Calling: For multiple samples, use
--snfoutput per sample, then combine withsniffles --input sample1.snf sample2.snf ... --vcf population.vcf.
Protocol 2: SV Calling with CuteSV from ONT Alignments
Objective: To sensitively detect structural variants using CuteSV's clustering algorithm.
Materials: As in Protocol 1, with CuteSV installed.
- Input Preparation: As in Protocol 1 (sorted, indexed BAM).
- Configuration File: Create a simple YAML config file (
cutesv_config.txt) to specify parameters: - SV Calling Execution:
./workdir: Temporary directory for intermediate files.--genotype: Enable genotyping of SVs.
- Output Filtering: CuteSV outputs raw calls. Consider filtering based on
RE(supporting reads) andAF(allele frequency) fields in the VCF for high-confidence sets.
Protocol 3: Consensus SV Set Generation
Objective: To generate a high-confidence SV callset by integrating results from Sniffles2 and CuteSV.
Materials: VCF outputs from Protocol 1 and Protocol 2.
- Normalization: Use
bcftools normto left-align and normalize indels in both VCFs against the reference. - Intersection: Use
SURVIVORorbcftools isecto find SVs called by both tools. Wheresample_files.txtlists the two VCF paths,1000is max distance between breakpoints,2indicates 2 callers,1 1 0specify require presence in at least 2 callers, type consistency, and no strand requirement. - Annotation: Annotate the consensus VCF with gene and repeat region information using tools like
SnpEfforAnnotSVfor biological interpretation.
Workflow and Relationship Diagrams
Title: SV Detection Workflow for ONT Data
Title: Core Logic of SV Calling Algorithms
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for SV Detection with ONT Reads
| Item | Function & Application |
|---|---|
| ONT Ligation Sequencing Kit (SQK-LSK114) | Prepares ultra-long genomic DNA libraries for sequencing. High molecular weight DNA input is critical for long-range SV detection. |
| NEB Monarch HMW DNA Extraction Kit | Extracts high molecular weight (>50 kbp) genomic DNA from tissues/cells, a prerequisite for generating ultra-long reads. |
| Genomic DNA Size Selection Beads (e.g., Circulomics SRE) | Performs size selection to enrich for the longest DNA fragments (>100 kbp), maximizing ultra-long read yield. |
| Reference Genome FASTA (e.g., GRCh38) | The reference sequence against which reads are aligned for SV detection. |
| Alignment Software (minimap2) | Aligns long, error-prone ONT reads to the reference genome, producing the BAM files used by SV callers. |
| SV Calling Software (Sniffles2, CuteSV) | Core analysis tools that detect structural variants from alignment patterns. |
| Benchmark Variant Sets (GIAB HG002) | Gold-standard truth sets for benchmarking SV call performance and tuning parameters. |
| Visualization Tool (IGV) | Allows visual inspection of read alignments and called SVs at candidate loci for validation. |
Ultra-long read (ULR) sequencing, primarily via Oxford Nanopore Technologies (ONT), is a transformative tool in genomics. This analysis, framed within a broader thesis on optimizing ONT ULR assembly workflows, delineates the specific scenarios where the benefits of ULRs justify their cost and technical demands over short-read (Illumina) and long-read (PacBio HiFi) technologies. The decision hinges on the biological complexity of the target and the specific genomic questions being asked.
Table 1: Comparative Overview of Major Sequencing Technologies (2024)
| Feature | Illumina (Short-Read) | PacBio HiFi (Long-Read) | ONT Ultra-Long (ULR) |
|---|---|---|---|
| Typical Read Length | 75-600 bp | 15-25 kb | 50 kb -> 1 Mb+ |
| Raw Read Accuracy | >99.9% (Q30) | >99.9% (Q20+) | ~95-98% (Q10-Q20) |
| Sequencing Chemistry | SBS (Synthesis) | SMRT (Circular Consensus) | Nanopore (Strand Sequencing) |
| Primary Strength | Cost-per-Gb, accuracy, high-throughput | High accuracy in long reads, SV detection | Maximum read length, direct epigenetic detection |
| Primary Limitation | Cannot resolve repeats/complex regions | Lower throughput, costlier than ONT standard | Higher DNA input/quality demands, lower base accuracy |
| Best Application | Variant calling, expression, resequencing | De novo assembly, haplotype phasing, SV in complex regions | Gapless T2T assemblies, complex SV, repetitive region resolution |
| Approx. Cost per Gb* | $5-$20 | $70-$120 | $15-$50 (standard), higher for ULR |
Costs are market estimates and vary by scale, region, and service model.
Table 2: Decision Matrix for Prioritizing ONT Ultra-Long Reads
| Research Goal | Recommended Technology | Rationale |
|---|---|---|
| Complete, gapless telomere-to-telomere (T2T) assembly | ONT ULR (Mandatory) | Reads span entire repetitive elements (e.g., centromeric satellite arrays) to provide unique overlaps. |
| Resolving mega-base pair structural variations (SVs) or complex haplotypes | ONT ULR (Highly Advantageous) | Single reads capture entire event architecture, eliminating assembly ambiguity. |
| High-quality de novo assembly of complex plant/animal genomes | PacBio HiFi or ONT ULR + Illumina | HiFi often provides better base accuracy; ONT ULR is superior if extreme repeats are the main barrier. |
| Targeted variant detection in a known genome | Illumina | Superior accuracy and cost-efficiency for known genomic contexts. |
| Direct detection of base modifications (e.g., 5mC) | ONT (Standard or ULR) | Native DNA sequencing enables direct epigenetic profiling. |
Detailed Experimental Protocol: ONT Ultra-Long Read DNA Extraction and Library Preparation
Protocol Title: High-Molecular-Weight (HMW) DNA Extraction and ULR Library Construction for ONT Sequencing.
Objective: To isolate ultra-high molecular weight (uHMW) DNA (>150 kb, N50 > 250 kb) and prepare a sequencing library optimized for ultra-long read generation on Oxford Nanopore PromethION or GridION platforms.
I. Materials & Reagent Solutions (The Scientist's Toolkit) Table 3: Key Research Reagent Solutions for ONT ULR Workflows
| Item | Function | Example Product/Note |
|---|---|---|
| Magen HMW Tissue DNA Kit | Isolation of intact, ultra-pure HMW DNA from cells/tissue. | Preferred for high yields and minimal shear. |
| Nanopore CS Kit (SQK-LSK114) | Recommended library prep kit for ULR sequencing. | Includes repair, end-prep, and ligation modules. |
| AMPure XP Beads | Size-selective purification and cleanup of DNA. | Critical for removing short fragments. |
| BluePippin or Short Read Eliminator (SRE) Kit | Size selection to enrich >50 kb fragments. | Essential for maximizing ULR output. |
| Qubit dsDNA HS Assay | Accurate quantification of low-concentration HMW DNA. | Fluorometric; more accurate than absorbance for HMW. |
| Pulse Field Gel Electrophoresis (PFGE) System | Quality assessment of DNA size distribution. | Gold-standard for visualizing uHMW DNA. |
| NEBNext FFPE DNA Repair Mix | Optional additional repair for challenging samples. | Enhances recovery of damaged DNA. |
II. Step-by-Step Protocol
A. uHMW DNA Extraction (from Cultured Cells)
- Cell Lysis: Harvest ~1-2 million cells. Embed cells in low-melt agarose plugs or use a gentle lysis buffer (from Magen kit) with extensive protease incubation (overnight at 50°C) to minimize shear.
- DNA Binding & Purification: Follow kit protocol. Use wide-bore or cut tips for all liquid transfers after lysis. Elute in a provided elution buffer or 10 mM Tris-HCl (pH 8.0). Do not vortex or centrifuge vigorously at any step.
- Quality Control:
- Quantification: Use Qubit. A260/A280 ratio should be ~1.8-2.0.
- Size Assessment: Analyze by PFGE or Femto Pulse system. Target N50 > 250 kb. A dense high-molecular-weight smear with minimal low-mass smear is ideal.
B. ONT ULR Library Preparation (SQK-LSK114)
- DNA Repair & End-Prep: Combine 3-7 µg of HMW DNA with repair components. Incubate at 20°C for 20 minutes, then 65°C for 20 minutes.
- Adapter Ligation: Add AMX adapter mix and Blunt/TA Ligase directly to the reaction. Incubate at room temperature for 30 minutes. Do not clean up the reaction.
- Size Selection (Critical Step): To the ligation mix, add 0.4x volume of AMPure XP beads to remove short fragments and excess adapters. Bind for 10 minutes, pellet, and discard supernatant. Wash beads twice with 70% ethanol. Elute DNA in Elution Buffer (EB).
- Optional Further Size Selection: For maximum ULR yield, use a BluePippin system with a 50 kb cutoff or a Short Read Eliminator Kit per manufacturer's instructions.
- Priming & Loading: Mix the eluted library with Sequencing Buffer (SB) and Loading Beads (LB). Load the entire volume onto a primed, fresh R10.4.1 or Q20+ flow cell.
C. Sequencing Run
- Start the sequencing run via MinKNOW software.
- Monitor initial pore activity and read N50 in real-time. A successful ULR run will show a gradually increasing read N50 over the first few hours.
- Run for up to 72 hours or until throughput plateaus.
Visualization: Decision Workflow and Experimental Pipeline
Decision Flowchart for Sequencing Technology Selection
ONT Ultra-Long Read Experimental Workflow
Conclusion
ONT ultra-long read assembly represents a paradigm shift in genomics, enabling the construction of complete, gapless, and haplotype-resolved genomes. This workflow, from foundational concepts through validation, empowers researchers to tackle previously intractable genomic regions, including centromeres, telomeres, and complex structural variants. For drug development and clinical research, these complete blueprints are invaluable for understanding genetic diversity, disease mechanisms, and regulatory landscapes. Future directions point toward real-time, on-device assembly for rapid pathogen characterization, integration with epigenomic data for functional insights, and the routine generation of phased diploid genomes as a new standard in personalized medicine. Mastering this workflow is now essential for any lab aiming to move beyond the limitations of short-read sequencing.