From Single Layers to Systems Biology: A Complete Guide to Modern Multi-Omics Analysis for Drug Discovery
This comprehensive guide introduces researchers, scientists, and drug development professionals to the integrated analysis of multi-omics data.
From Single Layers to Systems Biology: A Complete Guide to Modern Multi-Omics Analysis for Drug Discovery
Abstract
This comprehensive guide introduces researchers, scientists, and drug development professionals to the integrated analysis of multi-omics data. We begin by defining the core 'omics' layers—genomics, transcriptomics, proteomics, and metabolomics—and explaining the power of their integration for uncovering complex biological mechanisms. We then navigate through current methodologies, including batch effect correction, dimensionality reduction, and network analysis, with a focus on real-world applications in biomarker discovery and target identification. A dedicated section addresses common pitfalls in data integration, quality control, and statistical power, providing actionable troubleshooting strategies. Finally, we evaluate methods for validating multi-omics findings and comparing analysis tools. This article provides a foundational yet advanced roadmap for implementing robust multi-omics strategies to accelerate translational research.
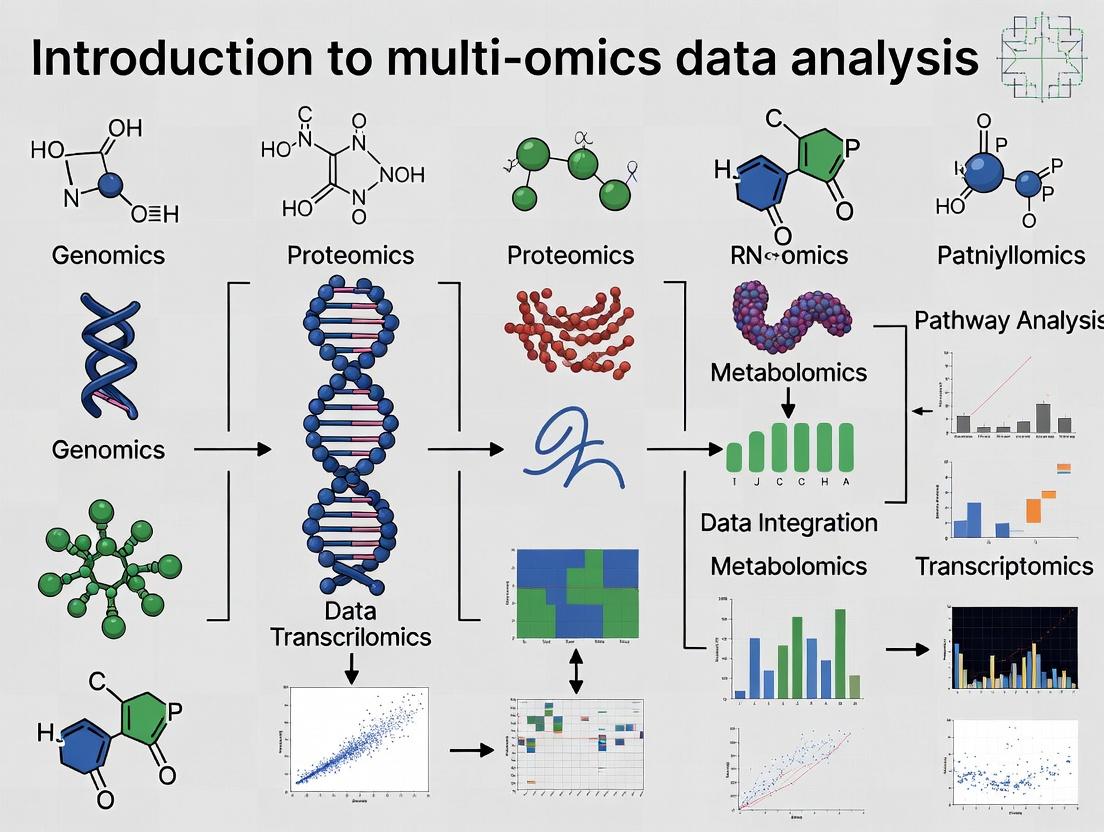
What is Multi-Omics? Demystifying Genomics, Transcriptomics, Proteomics, and Metabolomics for Systems Biology
The systematic analysis of biological systems requires an integrated approach beyond single data layers. This guide, framed within a broader thesis on Introduction to multi-omics data analysis research, details the core omics tiers—genomics, transcriptomics, proteomics, and metabolomics—that form the foundational data strata. Integration of these layers is essential for constructing comprehensive biological network models and identifying translatable biomarkers for complex disease and drug development.
The Hierarchical Omics Landscape: Core Definitions & Technologies
The flow of biological information from genotype to phenotype is captured through successive omics layers. Each layer employs distinct technologies for large-scale measurement.
Table 1: The Core Omics Tiers: Scope, Primary Technologies, and Output
| Omics Layer | Analytical Scope | Core Technology | Primary Output | Typical Sample Input |
|---|---|---|---|---|
| Genomics | DNA sequence, structure, variation | Next-Generation Sequencing (NGS), Microarrays | Sequence variants (SNPs, Indels), structural variants, epigenetic marks | DNA (genomic, bisulfite-treated) |
| Transcriptomics | RNA abundance & sequence | RNA-Seq, Microarrays, qRT-PCR | Gene expression levels, splice variants, non-coding RNA profiles | Total RNA, mRNA |
| Proteomics | Protein identity, quantity, modification | Mass Spectrometry (LC-MS/MS), Antibody Arrays | Protein identification, abundance, post-translational modifications (PTMs) | Proteins/Peptides (cell lysate, biofluid) |
| Metabolomics | Small-molecule metabolite profiles | Mass Spectrometry (GC-MS, LC-MS), NMR Spectroscopy | Metabolite identification and relative/absolute concentration | Serum, plasma, urine, tissue extract |
Detailed Experimental Protocols for Key Omics Analyses
Protocol: Whole-Transcriptome RNA Sequencing (RNA-Seq)
Objective: To profile the complete transcriptome, quantifying gene expression levels and identifying splice variants.
- RNA Extraction & QC: Isolate total RNA using TRIzol or silica-membrane kits. Assess integrity (RIN > 8.0) via Bioanalyzer.
- Library Preparation: Deplete ribosomal RNA or enrich poly-A mRNA. Fragment RNA, synthesize cDNA, and ligate sequencing adapters. Amplify via PCR.
- Sequencing: Load library onto an NGS platform (e.g., Illumina NovaSeq) for paired-end sequencing (e.g., 2x150 bp). Target 30-50 million reads per sample.
- Bioinformatics Analysis: Align reads to a reference genome (STAR, HISAT2). Quantify gene-level counts (featureCounts). Perform differential expression analysis (DESeq2, edgeR).
Protocol: Label-Free Quantitative Proteomics (LC-MS/MS)
Objective: To identify and quantify proteins in complex biological samples.
- Protein Extraction & Digestion: Lyse cells/tissue in RIPA buffer with protease inhibitors. Reduce (DTT), alkylate (IAA), and digest proteins with trypsin overnight at 37°C.
- Desalting: Desalt peptides using C18 solid-phase extraction (SPE) columns.
- LC-MS/MS Analysis: Separate peptides on a reverse-phase C18 nano-UHPLC column with a 60-90 min organic gradient. Analyze eluting peptides with a high-resolution tandem mass spectrometer (e.g., Q-Exactive) in data-dependent acquisition (DDA) mode.
- Data Processing: Identify proteins by searching MS/MS spectra against a protein database (MaxQuant, Proteome Discoverer). Quantify based on precursor ion intensity.
Protocol: Untargeted Metabolomics via LC-MS
Objective: To comprehensively profile small molecules in a biological sample.
- Metabolite Extraction: Add cold methanol/acetonitrile/water (4:4:2) to sample for protein precipitation. Vortex, centrifuge, and collect supernatant.
- LC-MS Analysis: Analyze in both positive and negative ionization modes. Use HILIC chromatography for polar metabolites and C18 for lipids. Employ a high-resolution mass spectrometer (e.g., Orbitrap) in full-scan mode (m/z 70-1000).
- Data Preprocessing: Perform peak picking, alignment, and annotation using software (XCMS, MS-DIAL). Annotate metabolites against public spectral libraries (mzCloud, GNPS).
- Statistical Analysis: Use multivariate analysis (PCA, PLS-DA) to identify differentially abundant metabolites.
Visualizing Omics Relationships and Workflows
Diagram 1: Central Dogma & Omics Flow
Diagram 2: Multi-Omics Analysis Pipeline
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Kits for Core Omics Workflows
| Item Name | Category | Function in Omics Research |
|---|---|---|
| QIAGEN DNeasy/RNeasy Kits | Genomics/Transcriptomics | Silica-membrane technology for high-purity, rapid isolation of genomic DNA or total RNA from various sample types. |
| Illumina TruSeq RNA Library Prep Kit | Transcriptomics | For preparation of stranded, paired-end RNA-seq libraries from total RNA, with mRNA enrichment or rRNA depletion. |
| Thermo Fisher Pierce BCA Protein Assay Kit | Proteomics | Colorimetric detection and quantification of total protein concentration, critical for sample normalization prior to MS analysis. |
| Trypsin, Sequencing Grade (Promega) | Proteomics | Protease for specific digestion of proteins at lysine/arginine residues, generating peptides for LC-MS/MS analysis. |
| C18 Solid-Phase Extraction (SPE) Cartridges | Metabolomics/Proteomics | Desalting and purification of peptides or metabolites from complex biological extracts prior to mass spectrometry. |
| Deuterated Internal Standards (e.g., CAMAG) | Metabolomics | Stable isotope-labeled compounds spiked into samples for quality control and to improve quantification accuracy in MS. |
| Bio-Rad Protease & Phosphatase Inhibitor Cocktails | General | Added to lysis buffers to prevent protein degradation and preserve post-translational modification states during extraction. |
In the field of multi-omics data analysis, a paradigm shift is underway from single-omics investigations to integrative approaches. This whitepaper posits that the strategic integration of genomics, transcriptomics, proteomics, and metabolomics data uncovers systemic biological insights that are fundamentally inaccessible through the analysis of any single layer in isolation. This emergent property—where the integrated whole is greater than the sum of its individual omics parts—is the Core Hypothesis of modern systems biology. We validate this through current evidence, provide a technical framework for integration, and outline its critical application in accelerating therapeutic discovery.
Quantitative Evidence for Integrative Superiority
Empirical studies consistently demonstrate that multi-omics integration yields a more complete and accurate picture of biological systems than unimodal analysis.
Table 1: Comparative Performance of Single vs. Multi-Omics Analyses in Disease Subtyping
| Study Focus | Single-Omics Approach (Best) | Classification Accuracy | Multi-Omics Integrated Approach | Classification Accuracy | Key Integrated Insight |
|---|---|---|---|---|---|
| Breast Cancer Subtypes | Transcriptomics (RNA-Seq) | 82-88% | RNA-Seq + DNA Methylation + miRNA | 94-97% | Revealed epigenetic drivers of transcriptional heterogeneity |
| Alzheimer's Disease Progression | Proteomics (Mass Spec) | 75-80% | GWAS + RNA-Seq + Proteomics + Metabolomics | 89-92% | Linked genetic risk loci to downstream metabolic pathway dysfunction |
| Colorectal Cancer Prognosis | Genomics (Mutation Panel) | 70-78% | WES + Transcriptomics + Immunohistochemistry | 91-95% | Identified immune-cold tumors masked by mutational load alone |
Table 2: Increase in Mechanistically Interpretable Findings from Integration
| Research Goal | Number of Significant Hits (Single-Omics) | Number of Significant Hits (Integrated) | Fold Increase | Nature of Gained Insights |
|---|---|---|---|---|
| Biomarker Discovery for NSCLC | 12 candidate proteins | 38 multi-omic features | 3.2x | Protein-metabolite complexes as superior early detectors |
| Pathway Elucidation in IBD | 3 dysregulated pathways | 11 coherent inter-omic pathways | 3.7x | Cascade from SNP->splicing->protein activity->metabolite output |
| Drug Target Prioritization | 5 high-interest genes | 15 ranked target modules | 3.0x | Contextualized druggable proteins within active network neighborhoods |
Foundational Methodologies for Multi-Omics Integration
Integration strategies are broadly categorized into a priori knowledge-driven and data-driven methods.
3.1 Early Integration (Data-Driven)
- Protocol: Concatenation-Based Fusion for Predictive Modeling
- Data Preprocessing: Independently normalize and scale each omics dataset (e.g., Z-score for RNA, Min-Max for methylation beta-values).
- Feature Reduction: Apply omics-specific dimensionality reduction (e.g., PCA on transcriptomics, UMAP on proteomics). Retain top components explaining >85% variance.
- Matrix Concatenation: Horizontally concatenate reduced matrices from n samples across m omics layers to form a unified feature matrix of size n x (p1+p2+...+pk).
- Joint Analysis: Feed the concatenated matrix into a machine learning model (e.g., Random Forest, Neural Network) for classification or regression.
- Validation: Use strict cross-validation where all data from a single patient is kept within the same fold to prevent data leakage.
3.2 Late Integration (Knowledge-Driven)
- Protocol: Pathway-Centric Integration Using Public Databases
- Individual Analysis: Perform differential expression/abundance analysis per omics layer. Generate lists of significant entities (e.g., genes, proteins, metabolites).
- Identifier Mapping: Map all entities to standard identifiers (e.g., Ensembl ID, Uniprot ID, HMDB ID) using tools like
g:ProfilerorMetaboAnalyst. - Pathway Enrichment: Conduct over-representation analysis (ORA) or gene set enrichment analysis (GSEA) per omics list against curated databases (KEGG, Reactome).
- Consensus Scoring: Integrate pathway scores using statistical meta-analysis methods (e.g., Fisher's combined probability test) or rank-aggregation (e.g., Robust Rank Aggregation).
- Causal Inference: Use prior knowledge graphs (e.g., from STRING, OmniPath) to infer directional flow from genomic variants to metabolomic changes, filling omics gaps with established interactions.
3.3 Intermediate/Hybrid Integration
- Protocol: Multi-Omics Factor Analysis (MOFA+)
- Data Input: Prepare omics datasets as a list of matrices, aligned by common samples.
- Model Training: Run MOFA+, a statistical framework that decomposes the data into a set of latent factors that capture shared and specific variations across omics types.
- Factor Interpretation: Correlate factors with sample covariates (e.g., disease status, survival) to interpret biological meaning.
- View-Specific Weights: Analyze the weight of each feature (gene, protein) in each factor per omics view to identify key drivers of inter-omic patterns.
Visualization of Core Integration Concepts
Multi-Omics Integration Reveals Latent Drivers
A Causally Linked Multi-Omics Cascade
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Platforms for Multi-Omics Research
| Category | Product/Platform Example | Core Function in Integration |
|---|---|---|
| Sample Prep (Nucleic Acids) | PAXgene Blood RNA/DNA System | Enables simultaneous stabilization of RNA and DNA from single blood sample, preserving molecular relationships. |
| Sample Prep (Proteins) | TMTpro 18-plex Isobaric Label Reagents | Allows multiplexed quantitative proteomics of up to 18 samples, directly aligning with transcriptomic cohorts. |
| Single-Cell Multi-Omics | 10x Genomics Multiome ATAC + Gene Expression | Profiles chromatin accessibility (ATAC) and transcriptome (RNA) from the same single nucleus. |
| Spatial Multi-Omics | NanoString GeoMx Digital Spatial Profiler | Enables region-specific, high-plex protein and RNA quantification from a single tissue section. |
| Mass Spectrometry | Thermo Scientific Orbitrap Astral Mass Spectrometer | Delivers deep-coverage proteomics and metabolomics, enabling direct correlation from a shared analytical platform. |
| Data Integration Software | QIAGEN OmicSoft Studio | Commercial platform for harmonizing, visualizing, and statistically analyzing disparate omics datasets. |
| Open-Source Analysis Suite | Snakemake or Nextflow Workflow Managers |
Orchestrates reproducible, modular pipelines for each omics type and their integration. |
Key Technologies and Platforms Generating Each Data Type (NGS, Mass Spectrometry, Arrays)
In the burgeoning field of multi-omics data analysis research, the integration of disparate biological data types is paramount for constructing a holistic understanding of complex systems. This technical guide details the core technologies and platforms responsible for generating the primary data types—from next-generation sequencing (NGS), mass spectrometry, and arrays—that form the foundation of genomics, proteomics, and metabolomics studies. A precise understanding of these data-generation engines is critical for designing robust integrative analyses in drug development and basic research.
Next-Generation Sequencing (NGS) Technologies
NGS platforms enable high-throughput, parallel sequencing of DNA and RNA, forming the bedrock of genomics and transcriptomics data.
Core Platforms & Technologies
| Platform (Vendor) | Core Technology | Key Output Data Type | Max Read Length | Throughput per Run (Approx.) | Primary Applications |
|---|---|---|---|---|---|
| NovaSeq X Series (Illumina) | Sequencing-by-Synthesis (SBS) with reversible terminators | Paired-end reads (FASTQ) | 2x 300 bp (X Plus) | Up to 16 Tb | Whole-genome, exome, transcriptome sequencing |
| Revio (PacBio) | Single Molecule, Real-Time (SMRT) Sequencing | HiFi reads (FASTQ) | 15-20 kb | 360 Gb | De novo assembly, variant detection, isoform sequencing |
| PromethION 2 (Oxford Nanopore) | Nanopore-based electronic sequencing | Long, direct reads (FAST5/FASTQ) | >4 Mb demonstrated | Up to 290 Gb | Ultra-long reads, real-time sequencing, direct RNA seq |
Detailed NGS Experimental Protocol: RNA-Sequencing
Objective: To generate a quantitative profile of the transcriptome. Key Reagents & Kits: See "The Scientist's Toolkit" below. Workflow:
- Total RNA Isolation: Extract RNA using guanidinium thiocyanate-phenol-chloroform (e.g., TRIzol) or column-based methods. Assess integrity (RIN > 8) via Bioanalyzer.
- Poly-A Selection or rRNA Depletion: Enrich for mRNA using oligo(dT) beads or remove ribosomal RNA using probe-based kits.
- cDNA Library Construction:
- Fragment RNA (or cDNA) to ~200-300 bp.
- Synthesize first-strand cDNA using reverse transcriptase and random hexamers/dT primers.
- Synthesize second-strand cDNA with DNA Polymerase I and RNase H.
- End-repair, A-tailing, and ligation of platform-specific adapters with sample indexes (barcodes).
- Library Amplification: Perform limited-cycle PCR to enrich for adapter-ligated fragments.
- Library QC: Quantify using fluorometry (Qubit) and assess size distribution (Bioanalyzer/TapeStation).
- Sequencing: Pool libraries at equimolar ratios and load onto the chosen NGS platform (e.g., Illumina NovaSeq) for cluster generation and sequencing-by-synthesis.
Diagram: Standard RNA-Seq Library Prep Workflow
Mass Spectrometry (MS) Platforms
MS platforms ionize and separate molecules based on their mass-to-charge ratio (m/z), generating proteomic and metabolomic data.
Core Platforms & Technologies
| Platform Category (Vendor Examples) | Ionization Source | Mass Analyzer(s) | Key Output Data Type | Key Applications |
|---|---|---|---|---|
| High-Resolution Tandem MS (Thermo Orbitrap Eclipse, Bruker timsTOF) | Electrospray (ESI), Nano-ESI | Quadrupole, Orbitrap, Time-of-Flight (TOF) | m/z spectra, fragmentation spectra (.raw, .d) | Discovery proteomics, phosphoproteomics, interactomics |
| MALDI-TOF/TOF (Bruker, SCIEX) | Matrix-Assisted Laser Desorption/Ionization (MALDI) | Time-of-Flight (TOF) | m/z peak lists | Microbial identification, imaging mass spec |
| GC-MS / LC-MS (Agilent, Waters) | EI/CI (GC), ESI (LC) | Quadrupole, Triple Quadrupole (QqQ) | Chromatograms & spectra | Targeted metabolomics, quantitation (MRM/SRM) |
Detailed MS Experimental Protocol: Bottom-Up Proteomics
Objective: To identify and quantify proteins in a complex sample. Key Reagents & Kits: See "The Scientist's Toolkit" below. Workflow:
- Protein Extraction & Quantification: Lyse cells/tissue in appropriate buffer (e.g., RIPA with protease inhibitors). Quantify via BCA or similar assay.
- Protein Digestion: Reduce disulfide bonds (DTT), alkylate cysteines (Iodoacetamide), and digest proteins into peptides using trypsin (typically overnight at 37°C).
- Peptide Cleanup/Desalting: Use C18 solid-phase extraction tips or columns to remove salts and detergents.
- Liquid Chromatography (LC): Separate peptides online via reversed-phase C18 column using a gradient of increasing organic solvent (acetonitrile).
- Mass Spectrometry Analysis (Data-Dependent Acquisition - DDA):
- Full MS Scan: The Orbitrap or TOF analyzer acquires a high-resolution MS1 spectrum.
- Peptide Selection: The most intense precursor ions are selected for fragmentation.
- Fragmentation: Selected ions are fragmented via Higher-energy Collisional Dissociation (HCD) or Collision-Induced Dissociation (CID).
- MS2 Scan: A high-resolution MS2 spectrum of the fragment ions is acquired.
- Data Output: Raw files containing paired MS1 and MS2 spectra for downstream database search.
Diagram: Bottom-Up Proteomics DDA Workflow
Array-Based Platforms
Arrays provide a high-throughput, multiplexed approach for profiling known targets via hybridization or affinity binding.
Core Platforms & Technologies
| Platform Type (Vendor Examples) | Core Technology | Key Output Data Type | Key Features | Primary Applications |
|---|---|---|---|---|
| Microarray (Affymetrix GeneChip, Agilent SurePrint) | Hybridization of labeled nucleic acids to immobilized probes | Fluorescence intensity data (.CEL, .GPR) | High multiplexing, cost-effective for known targets | Gene expression (mRNA, miRNA), SNP genotyping |
| Bead-Based Array (Illumina Infinium) | Hybridization to beads, followed by single-base extension | Fluorescence intensity data (.IDAT) | Scalable, high sample throughput | Methylation profiling (EPIC), GWAS |
| Protein/Antibody Array (RayBiotech, R&D Systems) | Affinity binding to immobilized antibodies or antigens | Chemiluminescence/fluorescence signals | Direct protein measurement, no digestion needed | Cytokine screening, phospho-protein profiling |
Detailed Array Experimental Protocol: Gene Expression Microarray
Objective: To measure the relative abundance of thousands of transcripts simultaneously. Key Reagents & Kits: See "The Scientist's Toolkit" below. Workflow:
- Total RNA Isolation & QC: As described in section 1.2. High-quality RNA is critical.
- cDNA Synthesis: Convert RNA into double-stranded cDNA using reverse transcriptase with a T7 promoter primer.
- cRNA Synthesis & Labeling: Perform in vitro transcription (IVT) from the cDNA template using T7 RNA polymerase and biotin- or Cy-labeled nucleotides to produce amplified, labeled cRNA.
- Fragmentation & Hybridization: Chemically fragment the labeled cRNA to uniform size and hybridize to the microarray under stringent conditions (16-20 hrs).
- Washing & Staining: Remove non-specifically bound material through a series of washes. Stain with a fluorescent conjugate (e.g., streptavidin-phycoerythrin for biotin) to detect bound target.
- Scanning & Data Acquisition: Scan the array with a confocal laser scanner to measure fluorescence intensity at each probe location.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item Name (Example Vendor) | Field of Use | Function & Brief Explanation |
|---|---|---|
| TRIzol Reagent (Thermo Fisher) | NGS / Arrays | A monophasic solution of phenol and guanidine isothiocyanate for simultaneous cell lysis and RNA/DNA/protein isolation. Denatures RNases. |
| NEBNext Ultra II DNA Library Prep Kit (NEB) | NGS | A comprehensive kit for converting DNA or RNA into sequencing-ready Illumina-compatible libraries, including fragmentation, end-prep, adapter ligation, and PCR modules. |
| Trypsin, Sequencing Grade (Promega) | Mass Spectrometry | A proteolytic enzyme that cleaves peptide bonds C-terminal to lysine and arginine residues, generating peptides of ideal size for MS analysis. |
| Pierce BCA Protein Assay Kit (Thermo Fisher) | Mass Spectrometry | A colorimetric assay based on bicinchoninic acid (BCA) for accurate colorimetric quantification of protein concentration. |
| GeneChip WT PLUS Reagent Kit (Thermo Fisher) | Arrays | Provides reagents for cDNA synthesis, IVT labeling, and fragmentation specifically optimized for Affymetrix whole-transcript expression arrays. |
| Hybridization Control Kit (CytoSure) | Arrays | Contains labeled synthetic oligonucleotides that bind to control spots on the array, allowing monitoring of hybridization efficiency and uniformity. |
Major Repositories and Public Databases for Multi-Omics Data (e.g., TCGA, GEO, PRIDE, Metabolomics Workbench)
The systematic integration of multiple molecular data layers—genomics, transcriptomics, proteomics, and metabolomics—is fundamental to modern systems biology and precision medicine. A critical first step in any multi-omics analysis research is the acquisition of high-quality, well-annotated public data. This guide provides an in-depth technical overview of the major repositories serving as the primary sources for such data, forming the empirical foundation upon which integrative computational analyses and biological discoveries are built.
Public data repositories are specialized archives designed to store, standardize, and disseminate large-scale omics data. They adhere to FAIR (Findable, Accessible, Interoperable, Reusable) principles and often require data submission as a condition of publication.
Table 1: Major Multi-Omics Data Repositories: Core Characteristics
| Repository Name | Primary Omics Focus | Data Types & Scope | Key Features & Standards | Access Method & Tools |
|---|---|---|---|---|
| The Cancer Genome Atlas (TCGA) | Genomics, Transcriptomics, Epigenomics | DNA-seq, RNA-seq, miRNA-seq, Methylation arrays, clinical data from ~33 cancer types. | Harmonized data via GDC; high-quality controlled pipelines; linked clinical outcomes. | GDC Data Portal, GDC API, TCGAbiolinks (R), GDC Transfer Tool. |
| Gene Expression Omnibus (GEO) | Transcriptomics, Epigenomics | Microarray, RNA-seq, ChIP-seq, methylation, and non-array data. Over 7 million samples. | MIAME/MINSEQE compliant; flexible platform; Series (study) and Sample-centric organization. | Web interface, GEO2R, GEOquery (R), SRA Toolkit for sequences. |
| Sequence Read Archive (SRA) | Genomics, Transcriptomics | Raw sequencing reads (NGS) from all technologies. Over 40 petabases of data. | Part of INSDC; stores raw data in FASTQ, aligned data in BAM/CRAM. | SRA Toolkit (prefetch, fasterq-dump), AWS/GCP buckets, ENA browser. |
| Proteomics Identifications (PRIDE) | Proteomics, Metabolomics (MS) | Mass spectrometry-based proteomics and metabolomics data: raw, processed, identification results. | MIAPE compliant; supports mzML, mzIdentML, mzTab; reanalysis via ProteomeXchange. | PRIDE Archive website, PRIDE API, PRIDE Inspector tool suite. |
| Metabolomics Workbench | Metabolomics | MS and NMR spectroscopy data from targeted and untargeted studies. Over 1,000 studies. | Supports a wide range of metabolomics data formats; detailed experimental metadata. | Web-based search, REST API, data download in various processed formats. |
| dbGaP | Genomics, Phenomics | Genotype-phenotype interaction studies. Includes GWAS, clinical, and molecular data. | Controlled-access for sensitive human data; strict protocols for data access approval. | Authorized access via eRA Commons; phenotype and genotype association browsers. |
| ArrayExpress | Transcriptomics, Epigenomics | Functional genomics data, primarily microarray and NGS-based. MIAME/MINSEQE compliant. | Curated data with ontology annotations; cross-references to ENA and PRIDE. | Web interface, API, R/Bioconductor packages. |
| GNPS (Global Natural Products Social Molecular Networking) | Metabolomics | Tandem mass spectrometry (MS/MS) data for natural products and metabolomics. | Enables molecular networking, spectral library matching, and repository-scale analysis. | Web platform, MASST search, Feature-Based Molecular Networking workflows. |
Table 2: Quantitative Summary of Repository Contents (Representative Stats)
| Repository | Estimated Studies | Estimated Samples/ Datasets | Primary Data Volume | Update Frequency |
|---|---|---|---|---|
| TCGA (via GDC) | 1 (pan-cancer program) | > 20,000 cases (multi-omic per case) | ~ 3.5 PB | Static, legacy archive. |
| GEO | > 150,000 | > 7,000,000 | Tens of PB | Daily submissions. |
| SRA | Millions of runs | > 40 Petabases of sequence data | > 40 PB | Continuous. |
| PRIDE | > 20,000 | > 1,000,000 datasets | ~ 1.5 PB | Weekly. |
| Metabolomics Workbench | > 1,200 | Not uniformly defined | ~ 50 TB | Regular submissions. |
Experimental Protocols and Data Generation Standards
The utility of public data hinges on the reproducibility of the underlying experiments. Below are generalized protocols for key omics technologies prevalent in these repositories.
Bulk RNA-Sequencing (Transcriptomics - representative for GEO, SRA, TCGA)
Protocol Title: Standard Workflow for Illumina Stranded Total RNA-Seq Library Preparation and Sequencing.
Key Steps:
- RNA Extraction & QC: Isolate total RNA using silica-membrane columns (e.g., RNeasy kit). Assess integrity via RIN (RNA Integrity Number) on Bioanalyzer. Require RIN > 7 for mammalian samples.
- rRNA Depletion: Use ribodepletion kits (e.g., Illumina Ribo-Zero Plus) to remove ribosomal RNA, enriching for mRNA and non-coding RNA.
- cDNA Synthesis & Library Prep: Fragment purified RNA. Synthesize first-strand cDNA with random hexamers and reverse transcriptase. Synthesize second strand incorporating dUTP for strand specificity. Perform end repair, A-tailing, and adapter ligation (using unique dual indices, UDIs).
- Library QC & Quantification: Purify libraries using SPRI beads. Quantify via fluorometry (Qubit). Assess size distribution via Bioanalyzer/Tapestation.
- Sequencing: Pool libraries equimolarly. Sequence on Illumina NovaSeq or HiSeq platform to a minimum depth of 30 million paired-end (2x150 bp) reads per sample.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) for Proteomics (representative for PRIDE)
Protocol Title: Data-Dependent Acquisition (DDA) Proteomics for Whole-Cell Lysate Analysis.
Key Steps:
- Protein Extraction & Digestion: Lyse cells in strong denaturant (e.g., 8M Urea, RIPA buffer). Reduce disulfide bonds with DTT (10mM, 30min, 56°C). Alkylate with iodoacetamide (25mM, 20min, dark). Digest with sequence-grade trypsin/Lys-C (1:50 enzyme:protein, 37°C, overnight) after dilution to 1-2M urea.
- Peptide Desalting: Desalt peptides using C18 solid-phase extraction (SPE) tips or StageTips. Elute with 60% acetonitrile/0.1% formic acid.
- LC-MS/MS Analysis: Reconstitute peptides in 0.1% formic acid. Load onto a C18 reverse-phase nanoLC column. Separate using a 60-180 min gradient from 2% to 35% acetonitrile. Interface with MS via nano-electrospray.
- Mass Spectrometry: Operate instrument (e.g., Q-Exactive HF, timsTOF) in DDA mode. Perform full MS1 scan (e.g., 60k resolution, 300-1750 m/z). Select top N most intense precursor ions (charge states 2-7) for fragmentation via higher-energy collisional dissociation (HCD). Acquire MS2 spectra at 15-30k resolution.
- Data Output: Raw instrument files (.raw, .d) are converted to open formats (.mzML) for submission.
Untargeted Metabolomics by LC-MS (representative for Metabolomics Workbench, GNPS)
Protocol Title: Global Metabolic Profiling of Plasma/Sera Using Reversed-Phase Chromatography and High-Resolution MS.
Key Steps:
- Sample Preparation: Deproteinize plasma/serum (e.g., 50 µL) with cold methanol or acetonitrile (1:4 ratio). Vortex, incubate at -20°C, centrifuge. Transfer supernatant and dry in a vacuum concentrator. Reconstitute in mobile phase starting conditions.
- Chromatographic Separation: Use a reversed-phase column (e.g., C18). Run a binary gradient: (A) Water + 0.1% formic acid; (B) Acetonitrile + 0.1% formic acid. Gradient from 2% B to 98% B over 15-25 minutes.
- Mass Spectrometry in Polarity Switching Mode: Use a high-resolution Q-TOF or Orbitrap mass spectrometer. Acquire data in both positive and negative electrospray ionization (ESI+/-) modes alternately. Acquire full-scan data at high resolution (> 50,000 FWHM) with a mass range of ~50-1200 m/z.
- Data-Dependent MS/MS: In parallel, acquire fragmentation spectra for top ions in each scan cycle to generate experimental MS/MS spectral libraries.
- Quality Controls: Inject pooled quality control (QC) samples repeatedly throughout the batch to monitor instrument stability.
Visualizations of Data Flows and Relationships
Title: Multi-Omics Data Lifecycle from Sample to Repository
Title: Multi-Omics Analysis Workflow from Repositories to Insight
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Multi-Omics Data Generation
| Item Name | Vendor Examples | Function in Protocol |
|---|---|---|
| RNeasy Mini/Midi Kit | Qiagen | Silica-membrane based purification of high-quality total RNA from various samples; critical for transcriptomics. |
| KAPA HyperPrep Kit | Roche | A widely used library preparation kit for Illumina sequencing from DNA or RNA, offering robust performance for genomics/transcriptomics. |
| Illumina Stranded Total RNA Prep with Ribo-Zero Plus | Illumina | Integrated kit for ribodepletion and stranded RNA-seq library construction, ensuring comprehensive transcriptome coverage. |
| Trypsin/Lys-C Mix, Mass Spec Grade | Promega | Proteolytic enzyme for specific digestion of proteins into peptides; gold standard for bottom-up proteomics. |
| S-Trap or FASP Columns | Protifi, Expedeon | Filter-aided or column-based devices for efficient protein digestion and cleanup, ideal for detergent-containing lysates. |
| Pierce C18 Spin Tips | Thermo Fisher Scientific | For desalting and concentrating peptide samples prior to LC-MS/MS analysis, improving sensitivity. |
| Mass Spectrometry Internal Standards Kit | Cambridge Isotope Labs | Stable isotope-labeled compounds added to metabolomics samples for quality control and semi-quantitative analysis. |
| Bioanalyzer RNA Nano or High Sensitivity Kits | Agilent | Microfluidics-based electrophoresis for precise assessment of RNA or DNA library quality and quantity. |
| Qubit dsDNA HS/RNA HS Assay Kits | Thermo Fisher Scientific | Fluorometric quantification of nucleic acids, offering high specificity over spectrophotometric methods. |
| Unique Dual Index (UDI) Kits | Illumina, IDT | Oligonucleotide sets for multiplexing samples, ensuring accurate sample demultiplexing and reducing index hopping artifacts. |
The integration of multi-omics data (genomics, transcriptomics, proteomics, metabolomics) is central to modern systems biology. Effective visualization is not merely illustrative but an analytical tool for hypothesis generation, pattern recognition, and communicating complex biological narratives. This guide details three pivotal visualization techniques within the context of a multi-omics analysis research framework.
Core Visualization Techniques: Methods and Applications
Heatmaps: For Pattern Discovery and Clustering
Methodology: Heatmaps are matrix representations where individual values are colored. In multi-omics, they are essential for visualizing gene expression (RNA-seq), protein abundance, or metabolite levels across multiple samples.
- Data Normalization: Apply Z-score normalization (for rows/features) or log2 transformation (for count data like RNA-seq) to make values comparable.
- Clustering: Perform hierarchical clustering (using Euclidean or correlation distance and Ward's or average linkage) on both rows (features) and columns (samples) to group similar patterns.
- Color Scaling: Choose a diverging colormap (e.g., blue-white-red) for Z-scores or a sequential colormap (e.g., white to dark blue) for normalized abundance.
- Annotation: Add side-columns to annotate sample groups (e.g., disease vs. control) or feature metadata (e.g., gene pathway).
Table 1: Common Clustering & Distance Metrics for Heatmaps
| Aspect | Option 1 | Option 2 | Use Case |
|---|---|---|---|
| Distance Metric | Euclidean Distance | Pearson Correlation | Euclidean for absolute magnitude, Correlation for pattern shape. |
| Linkage Method | Ward's Method | Average Linkage | Ward's minimizes variance; Average is less sensitive to outliers. |
| Normalization | Row Z-score | Log2(CPM+1) | Z-score for relative change; Log-CPM for sequencing count data. |
Methodology: Circos plots display connections between genomic loci or data tracks in a circular layout, ideal for showing structural variants, copy number variations, or correlations between different omics layers on a chromosomal scale.
- Data Preparation: Format data into tracks (e.g., ideogram, scatter plot, histogram, link). Each track requires genomic coordinates (chromosome, start, end).
- Ideogram Setup: Define chromosomes as the plot's backbone using a genome reference file (e.g., hg38).
- Adding Tracks: Plot quantitative data (e.g., gene expression fold-change) as scatter points or histograms on outer tracks.
- Drawing Links: Represent relationships (e.g., fusion genes, chromatin interactions) as ribbons or lines connecting two genomic regions. Link thickness can encode a value like read pair support.
Pathway & Network Diagrams: For Functional Interpretation
Methodology: These diagrams contextualize omics data within biological pathways (e.g., KEGG, Reactome) or protein-protein interaction networks, translating gene lists into mechanistic insights.
- Overlay Data: Map differentially expressed genes or altered proteins onto a canonical pathway. Use a continuous color gradient on node symbols to represent fold-change or p-value.
- Enrichment Visualization: Create bubble charts or bar graphs where node size = gene count, color = enrichment p-value, and position groups related pathways.
- Custom Network Building: Use interaction databases (STRING, BioGRID) to build networks from significant hits, then apply layout algorithms (force-directed, circular) for clarity.
Table 2: Key Reagents & Tools for Multi-Omics Visualization
| Item / Resource | Function / Purpose |
|---|---|
| R/Bioconductor | Primary platform for statistical analysis and generation of publication-quality heatmaps (pheatmap, ComplexHeatmap) and Circos plots (circlize). |
| Python (Matplotlib, Seaborn, Plotly) | Libraries for creating interactive and static visualizations, including advanced heatmaps and network graphs. |
| Cytoscape | Standalone software for powerful, customizable network visualization and analysis, especially for pathway diagrams. |
| Adobe Illustrator / Inkscape | Vector graphics editors for final polishing, annotation, and layout adjustment of figures for publication. |
| KEGG / Reactome / WikiPathways | Databases providing curated pathway maps in standardized formats (KGML, SBGN) for data overlay. |
| UCSC Genome Browser / IGV | Reference tools for visualizing genomic coordinates and aligning custom tracks, informing Circos plot design. |
Experimental Protocol: Integrative Multi-Omics Analysis Workflow
This protocol outlines a standard pipeline for generating data suitable for the visualizations described.
Title: Differential Analysis of Transcriptome and Proteome in Treatment vs. Control Cell Lines.
- Sample Preparation:
- Culture cells in triplicate for treated and control conditions.
- Harvest cells, divide aliquots for RNA and protein extraction.
- Multi-Omics Data Generation:
- RNA-seq: Extract total RNA (QIAGEN RNeasy), assess quality (RIN > 8, Bioanalyzer). Prepare libraries (Illumina TruSeq Stranded mRNA), sequence on NovaSeq (2x150bp).
- Proteomics (LC-MS/MS): Lyse protein pellets, digest with trypsin, label with TMTpro 16-plex reagents. Fractionate by high-pH reverse-phase HPLC, analyze on Orbitrap Eclipse.
- Bioinformatics Processing:
- Transcriptomics: Align reads to reference genome (hg38) with STAR. Quantify gene counts with featureCounts. Perform differential expression with
DESeq2(FDR < 0.05, |log2FC| > 1). - Proteomics: Process raw files with MaxQuant. Search against human UniProt database. Perform differential abundance analysis with
limmaon log2-transformed TMT intensities.
- Transcriptomics: Align reads to reference genome (hg38) with STAR. Quantify gene counts with featureCounts. Perform differential expression with
- Integrative Visualization:
- Heatmap: Create a unified heatmap of significant genes/proteins (Z-scores) across all samples.
- Pathway Analysis: Perform GSEA on both datasets. Visualize enriched pathways (e.g., "Apoptosis Signaling") as annotated diagrams.
- Circos Plot: Generate a plot showing chromosomal locations of key dysregulated genes and proteins, with links indicating cis-correlations.
Diagram: Multi-Omics Data Analysis & Visualization Workflow
Diagram: Key Immune Signaling Pathway (NF-κB)
Multi-Omics Integration in Action: Step-by-Step Workflows, Tools, and Applications in Biomarker Discovery
Multi-omics integrates diverse biological data sets—including genomics, transcriptomics, proteomics, and metabolomics—to construct a comprehensive model of biological systems. Framed within a broader thesis on introduction to multi-omics data analysis research, this technical guide provides a high-level overview of the end-to-end pipeline, from raw data generation to functional biological insight, targeting researchers and drug development professionals.
The canonical pipeline consists of four sequential, interconnected stages: Data Generation & Processing, Multi-Omics Integration, Biological Interpretation, and Validation & Insight.
Diagram Title: Multi-Omics Pipeline Core Stages
Stage 1: Data Generation & Processing
This stage involves converting biological samples into quantitative digital data. Each omics layer requires specific experimental and computational protocols.
Table 1: Core Omics Layers & Data Processing Tools
| Omics Layer | Core Technology | Primary Output | Key Processing Tools (Examples) | Typical Data Matrix |
|---|---|---|---|---|
| Genomics | Next-Generation Sequencing (NGS) | FASTQ files | BWA, GATK, SAMtools | Variant Call Format (VCF) |
| Transcriptomics | RNA-Seq, Microarrays | FASTQ or .CEL files | STAR, HISAT2, DESeq2, limma | Gene Expression Counts/FPKM |
| Proteomics | Mass Spectrometry (LC-MS/MS) | .raw spectra files | MaxQuant, MSFragger, DIA-NN | Peptide/Protein Abundance |
| Metabolomics | LC/GC-MS, NMR | .raw spectra files | XCMS, MS-DIAL, MetaboAnalyst | Metabolite Abundance |
Detailed Protocol: RNA-Seq Data Processing (Example)
- Quality Control (QC): Use FastQC to assess read quality. Trim adapters and low-quality bases with Trimmomatic or Cutadapt.
- Alignment: Map reads to a reference genome (e.g., GRCh38) using a splice-aware aligner like STAR. Output: BAM files.
- Quantification: Count reads mapping to genomic features (genes, exons) using featureCounts or HTSeq. Generate a counts matrix.
- Normalization & Differential Expression: Import counts into R/Bioconductor. Use DESeq2 to normalize for library size and composition, then perform statistical testing to identify differentially expressed genes (FDR < 0.05).
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Multi-Omics |
|---|---|
| Poly(A) mRNA Magnetic Beads | Isolates eukaryotic mRNA from total RNA for RNA-Seq library prep. |
| Trypsin (Sequencing Grade) | Digests proteins into peptides for bottom-up LC-MS/MS proteomics. |
| TMT/Isobaric Tags | Allows multiplexed quantification of up to 16 samples in a single MS run. |
| Methanol (LC-MS Grade) | Extracts and preserves metabolites for metabolomics; high purity prevents ion suppression. |
| KAPA HyperPrep Kit | Robust library preparation kit for NGS, compatible with degraded inputs. |
| Phosphatase/Protease Inhibitors | Preserves post-translational modification states in proteomics samples. |
Stage 2: Multi-Omics Data Integration
Integration methods correlate features across omics layers to identify master regulators and unified signatures.
Table 2: Common Multi-Omics Integration Methods
| Method Type | Description | Key Algorithms/Tools | Use Case |
|---|---|---|---|
| Concatenation-Based | Merges datasets into a single matrix for joint analysis. | MOFA, DIABLO | Identifying multi-omics biomarkers for patient stratification. |
| Network-Based | Constructs correlation or regulatory networks. | WGCNA, miRLAB | Inferring gene-metabolite interaction networks. |
| Similarity-Based | Integrates via kernels or statistical similarity. | Similarity Network Fusion (SNF) | Cancer subtype discovery from complementary data. |
| Model-Based | Uses statistical models to infer latent factors. | MOFA, Integrative NMF | Deconvolving shared vs. dataset-specific variations. |
Diagram Title: Multi-Omics Data Integration Approaches
Stage 3: Biological Interpretation & Pathway Analysis
Integrated features are mapped to biological knowledge bases for functional insight.
Detailed Protocol: Overrepresentation Analysis (ORA)
- Input: A list of significant integrated features (e.g., genes and metabolites).
- Background Definition: Define the set of all features measured in the experiment.
- Statistical Test: Use a hypergeometric test or Fisher's exact test to assess if features from a specific pathway (e.g., from KEGG, Reactome) appear in your list more often than expected by chance.
- Multiple Testing Correction: Apply Benjamini-Hochberg procedure to control the False Discovery Rate (FDR). Pathways with FDR < 0.05 are considered significantly enriched.
Diagram Title: Biological Interpretation from Integrated Signature
Stage 4: Validation & Translational Insight
Hypotheses generated in silico must be validated experimentally. Common approaches include:
- Targeted Assays: Using qPCR (genes), Western Blot/SRM (proteins), or targeted MS (metabolites) to confirm key findings in a new sample set.
- Functional Experiments: In vitro (knockdown/overexpression in cell lines) or in vivo studies to establish causal relationships.
- Clinical Correlation: Validating multi-omics biomarkers against patient outcomes in independent cohorts.
The final output is refined biological insight, which may include novel therapeutic targets, diagnostic biomarkers, or an advanced understanding of disease mechanisms, directly informing drug development pipelines.
Data Preprocessing and Normalization Strategies for Heterogeneous Datasets
Within the context of multi-omics data analysis research, the integration of heterogeneous datasets—spanning genomics, transcriptomics, proteomics, and metabolomics—presents a formidable challenge. Each omics layer is generated via distinct technologies, resulting in data with varying scales, distributions, missingness, and noise profiles. Effective preprocessing and normalization are not merely preliminary steps but are foundational to deriving biologically meaningful and statistically robust integrated models. This guide details current strategies to transform raw, disparate data into a coherent analytical framework.
Core Preprocessing Challenges in Multi-Omics Data
Each data type requires specific handling before cross-omics normalization can occur.
Table 1: Characteristic Challenges by Omics Data Type
| Data Type | Typical Format | Key Preprocessing Needs | Common Noise Sources |
|---|---|---|---|
| Genomics (e.g., SNP) | Variant counts/calls | Quality score filtering, linkage disequilibrium pruning, imputation. | Sequencing errors, batch effects. |
| Transcriptomics | RNA-seq read counts | Adapter trimming, quality control, alignment, count generation. | Library size, GC content, ribosomal RNA. |
| Proteomics | Mass spectrometry peaks | Peak detection/alignment, background correction, ion current normalization. | Ion suppression, instrument drift. |
| Metabolomics | NMR/LC-MS spectral peaks | Spectral alignment, baseline correction, solvent peak removal. | Matrix effects, day-to-day variability. |
Experimental Protocols for Key Preprocessing Steps
Protocol 3.1: RNA-seq Read Normalization (DESeq2 Median-of-Ratios Method)
- Input: Raw count matrix (genes x samples).
- Step 1 - Calculate gene-wise geometric mean: For each gene, compute the geometric mean of counts across all samples.
- Step 2 - Calculate sample-wise ratios: For each sample, divide each gene's count by the gene's geometric mean (creating a ratio). Genes with a geometric mean of zero or ratios in the extreme upper/lower quantiles are excluded.
- Step 3 - Derive size factor: The size factor for each sample is the median of its non-excluded gene ratios.
- Step 4 - Normalize: Divide the raw counts for each sample by its calculated size factor.
- Output: Normalized count matrix suitable for between-sample comparison.
Protocol 3.2: Probabilistic Quotient Normalization (PQN) for Metabolomics
- Input: Pre-aligned spectral intensity matrix (features x samples).
- Step 1 - Select Reference: Calculate the median spectrum (feature-wise median across all samples) as the reference.
- Step 2 - Calculate Quotients: For each sample, compute the quotient of each feature's intensity divided by the corresponding reference intensity.
- Step 3 - Determine Dilution Factor: Calculate the median of all quotients for that sample. This is the estimated dilution factor.
- Step 4 - Normalize: Divide all feature intensities in the sample by its dilution factor.
- Output: Concentration-corrected intensity matrix, reducing urine/serum dilution variability.
Normalization Strategies for Dataset Integration
Post individual-layer preprocessing, strategies to enable cross-omics analysis are applied.
Table 2: Cross-Platform Normalization Strategies
| Strategy | Principle | Best For | Key Limitation |
|---|---|---|---|
| Quantile Normalization | Forces all sample distributions (per platform) to be identical. | Technical replicate harmonization. | Removes true biological inter-sample variance. |
| ComBat / limma | Empirical Bayes framework to adjust for known batch effects. | Removing strong, known batch covariates. | Requires careful model specification. |
| Mean-Centering & Scaling (Auto-scaling) | Subtract mean, divide by standard deviation per feature. | Making features unit variance for downstream ML. | Amplifies noise in low-variance features. |
| Domain-Specific Normalization | Apply optimal single-omics method (e.g., DESeq2 for RNA-seq, PQN for metabolomics) separately before concatenation. | Preserving data-type-specific biological signals. | Does not correct for inter-omics scale differences. |
| Singular Value Decomposition (SVD) | Removes dominant orthogonal components assumed to represent technical noise. | Unsupervised batch effect removal. | Risk of removing biologically relevant signal. |
Visualization of Workflows and Relationships
Multi-Omics Data Preprocessing and Normalization Pipeline
Decision Guide for Selecting a Normalization Strategy
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Multi-Omics Preprocessing
| Item / Reagent | Function in Preprocessing/Normalization |
|---|---|
| FastQC / MultiQC | Quality control software for sequencing and array data; aggregates reports across samples and omics layers. |
| Trim Galore! / Trimmomatic | Removes adapter sequences and low-quality bases from NGS reads, critical for accurate alignment. |
| DESeq2 (R/Bioconductor) | Performs median-of-ratios normalization and differential expression analysis for count-based RNA-seq data. |
| limma (R/Bioconductor) | Applies linear models to microarray or RNA-seq data for differential expression and batch effect removal. |
| ComBat (sva R package) | Empirical Bayes method to adjust for batch effects in high-dimensional data across platforms. |
| MetaboAnalyst | Web-based platform offering multiple normalization protocols (e.g., PQN, sample-specific) for metabolomics. |
| SIMCA-P+ / Eigenvector Solo | Commercial software with advanced tools for multiplicative scatter correction (MSC) in spectral data. |
| Python Scikit-learn | Provides StandardScaler, RobustScaler, and Normalizer classes for feature-wise scaling post-integration. |
Within the burgeoning field of multi-omics data analysis research, the integration of disparate biological data layers—such as genomics, transcriptomics, proteomics, and metabolomics—is paramount for constructing a holistic understanding of complex biological systems and disease mechanisms. This technical guide details four core methodological paradigms for multi-omics integration: Concatenation, Correlation, Network, and Machine Learning-Based methods. Each approach presents unique advantages, challenges, and appropriate contexts for application, directly supporting the central thesis that sophisticated integration is the key to unlocking translational insights in biomedical research and drug development.
Concatenation-Based Methods
Concatenation, or early integration, involves merging raw or processed data matrices from multiple omics layers into a single, combined matrix prior to analysis.
Methodology
The core protocol involves:
- Data Preprocessing & Normalization: Each omics dataset is independently normalized (e.g., using variance stabilizing transformation for RNA-seq, quantile normalization for microarrays, or probabilistic quotient normalization for metabolomics) and scaled (e.g., Z-score) to ensure comparability across features with vastly different dynamic ranges.
- Feature Space Union: The normalized matrices are joined horizontally (sample-wise) or vertically (feature-wise). The most common approach is sample-wise concatenation, creating a unified matrix
X_integratedof dimensions(n_samples, n_features_omics1 + n_features_omics2 + ...). - Dimensionality Reduction & Analysis: The high-dimensional concatenated matrix is subjected to multivariate analysis. Principal Component Analysis (PCA) or Multiple Factor Analysis (MFA) are frequently employed to project the data into a lower-dimensional space where samples can be visualized and clusters identified.
Table 1: Quantitative Comparison of Key Concatenation Analysis Tools
| Tool/Method | Key Algorithm | Input Data Type | Primary Output | Typical Runtime for N=100, p=10k |
|---|---|---|---|---|
| MOFA+ | Factor Analysis | Multi-modal matrices | Latent factors, weights | ~10-30 minutes |
| Multiple Factor Analysis (MFA) | Generalized PCA | Quantitative matrices | Combined sample factors | <5 minutes |
| iCluster | Joint Latent Variable | Discrete/Continuous | Integrated clusters | ~15-60 minutes |
Multi-Omics Concatenation and Analysis Workflow
Research Reagent Solutions
- Benchmarking Datasets: Pre-processed, gold-standard multi-omics datasets (e.g., TCGA Pan-Cancer, CPTAC) are essential for validating concatenation pipelines.
- Normalization Reagents: Spike-in controls (e.g., SIRMs for metabolomics, ERCC RNA spikes for transcriptomics) are critical for cross-platform normalization.
- Integrated Analysis Software: Licensed software like SIMCA-P (for MFA) or dedicated R/Python packages (e.g.,
mointegrator,omicade4) provide the computational environment.
Correlation-Based Methods
Correlation, or pairwise integration, identifies statistical relationships between features across different omics datasets, often measured on the same samples.
Methodology
A standard protocol for cross-omics correlation analysis:
- Dataset Preparation: Generate paired datasets where
X(e.g., mRNA expression, dimensions n x p) andY(e.g., protein abundance, dimensions n x q) are measured from the samenbiological samples. - Correlation Matrix Computation: Calculate all pairwise correlations between features in
XandY. Common metrics include Pearson'sr(for linear relationships), Spearman'sρ(for monotonic), or sparse canonical correlation analysis (sCCA) for high-dimensional data. - Statistical Inference & Multiple Testing Correction: Assess the significance of each correlation (e.g., via t-test for Pearson's r) and apply corrections (Benjamini-Hochberg FDR) to control false discoveries.
- Biological Interpretation: Significant cross-omics feature pairs (e.g., gene-protein) are mapped to pathways (KEGG, Reactome) using enrichment analysis tools.
Cross-Omics Correlation Analysis Pipeline
Network-Based Methods
Network approaches model biological systems as graphs, where nodes represent biomolecules from various omics layers and edges represent functional or physical interactions.
Experimental Protocol
Protocol for Multi-Layer Network Construction:
- Layer-Specific Network Inference: Construct individual omics networks (e.g., gene co-expression via WGCNA, protein-protein interaction from STRING).
- Integration via Similarity or Propagation: Fuse networks using methods like Similarity Network Fusion (SNF):
a. For each omics data type, construct a sample similarity network (affinity matrix
W). b. Normalize each network:P = D^{-1} W, whereDis the diagonal degree matrix. c. Iteratively update each network using the formula:P^{(v)} = S^{(v)} * ( (∑_{k≠v} P^{(k)}) / (V-1) ) * (S^{(v)})^T, whereS^{(v)}is the similarity for viewv, fortiterations. d. Fuse the stabilized networks:P_{fused} = (1/V) ∑_{v=1}^{V} P^{(v)}. - Cluster Detection & Analysis: Perform spectral clustering on
P_{fused}to identify multi-omics patient subtypes. Analyze differential features across clusters.
Table 2: Network Integration Tools and Performance
| Tool | Integration Strategy | Network Types Supported | Key Output | Scalability (Max Samples) |
|---|---|---|---|---|
| Similarity Network Fusion (SNF) | Iterative Message Passing | Sample similarity | Fused network, clusters | ~1,000 |
| MOGAMUN | Multi-Objective Genetic Algorithm | PPI + Expression | Subnetworks | ~500 genes |
| OmicsIntegrator | Prize-Collecting Steiner Forest | PPI + any omics | Context-specific networks | ~10,000 nodes |
Multi-Layer Network Fusion and Clustering
Research Reagent Solutions
- Reference Interaction Databases: Curated knowledge bases (e.g., STRING, BioGRID, Recon3D metabolic model) serve as scaffold networks.
- Network Visualization Software: Tools like Cytoscape with dedicated plugins (Omics Visualizer, enhancedGraphics) are mandatory for interpretation.
- High-Performance Computing (HPC) Resources: Network algorithms are computationally intensive, requiring access to HPC clusters with adequate RAM (≥64 GB) and multi-core processors.
Machine Learning-Based Methods
ML methods, particularly supervised and deep learning models, learn complex, non-linear patterns from integrated omics data for predictive modeling.
Detailed Methodology
Protocol for a Deep Learning-Based Multi-Omics Classifier (e.g., for Disease Prediction):
- Data Partitioning & Input Engineering: Split samples into training (70%), validation (15%), and test (15%) sets. For each omics type, design an input encoding layer (e.g., a dense layer for molecular features).
- Model Architecture Definition: Implement a multi-modal neural network.
- Input Layers: Separate input tensors for each omics type.
- Encoder Branches: Each branch contains dense layers with batch normalization, ReLU activation, and dropout (e.g., 0.5) for feature extraction.
- Integration Layer: Concatenate the outputs of all branches.
- Classifier Head: Dense layers culminating in a softmax output for classification.
- Model Training & Validation: Train using the Adam optimizer with a categorical cross-entropy loss. Monitor validation loss for early stopping. Use gradient-based attribution methods (e.g., SHAP, Integrated Gradients) on the trained model to identify influential features across omics layers.
Table 3: Comparison of ML Integration Approaches
| Method Class | Example Algorithms | Handles High Dimensionality | Models Non-linearity | Interpretability |
|---|---|---|---|---|
| Supervised (Late Integration) | Stacked Generalization, MOFA + Classifier | Moderate | Yes | Moderate |
| Deep Learning (Hybrid) | Multi-modal Autoencoders, DeepION | Yes (with regularization) | High | Low (requires XAI) |
| Kernel Methods | Multiple Kernel Learning (MKL) | Yes | Yes | Low |
Deep Learning Model for Multi-Omics Integration
Research Reagent Solutions
- Curated Benchmark Suites: Frameworks like
MultiBenchprovide standardized datasets and protocols for fair ML model comparison. - Explainable AI (XAI) Tools: Software libraries (e.g.,
SHAP,Captum,LIME) are indispensable for interpreting "black-box" model predictions. - Specialized ML Platforms: Cloud-based AI platforms (Google Vertex AI, NVIDIA CLARA) offer optimized environments for developing and deploying large multi-omics models.
The selection of a multi-omics integration approach—concatenation, correlation, network, or machine learning—is contingent upon the specific biological question, data characteristics, and desired outcome. Concatenation and correlation offer intuitive starts, while network and ML methods provide powerful, albeit complex, frameworks for uncovering deep biological insights. As the field matures, hybrid methods that combine the strengths of these paradigms will be central to advancing the thesis of multi-omics research, ultimately accelerating biomarker discovery and therapeutic development.
Essential Software and R/Python Packages (e.g., MixOmics, MOFA, OmicsPlayground)
Multi-omics data integration is a cornerstone of modern systems biology, enabling researchers to derive a holistic understanding of biological systems. This guide, framed within a broader thesis on multi-omics data analysis, provides an in-depth technical overview of essential software and packages for researchers, scientists, and drug development professionals. We focus on three pivotal tools: MixOmics, MOFA, and OmicsPlayground.
Core Packages and Software
Quantitative Comparison of Core Tools
The following table summarizes key quantitative and functional attributes of the featured tools.
Table 1: Comparison of Multi-Omics Integration Tools
| Feature | MixOmics (R) | MOFA (R/Python) | OmicsPlayground (R/Web) |
|---|---|---|---|
| Primary Method | Projection (PLS, sPLS, DIABLO) | Factor Analysis (Bayesian) | Exploratory Analysis & Visualization Suite |
| Omics Types Supported | Transcriptomics, Metabolomics, Proteomics, Microbiome | Any (Designed for heterogeneous data) | Transcriptomics, Proteomics, Metabolomics, Single-cell |
| Key Strength | Dimensionality reduction, supervised integration | Unsupervised discovery of latent factors | Interactive GUI, no-code analysis, extensive preprocessing |
| Integration Model | Multi-block, multivariate | Statistical, factor-based | Modular, workflow-based |
| Typical Output | Component plots, loadings, network inferences | Factor values, weights, variance decomposition | Interactive plots, biomarker lists, pathway maps |
| Best For | Class prediction, biomarker discovery, correlation | Uncovering hidden sources of variation across datasets | Rapid hypothesis generation, data exploration, validation |
| License | GPL-2/3 | LGPL-3 | Freemium (Academic/Commercial) |
| Latest Version (as of 2024) | 6.24.0 | 2.0 (MOFA2) / 1.6.0 (MOFA+) | 3.0 |
The Scientist's Toolkit: Research Reagent Solutions
This table details essential computational "reagents" for conducting multi-omics integration studies.
Table 2: Essential Research Reagent Solutions for Multi-Omics Analysis
| Item | Function/Explanation |
|---|---|
| High-Performance Compute (HPC) Cluster or Cloud Credits | Essential for running resource-intensive integration algorithms and large-scale permutations. |
| Curated Reference Databases (e.g., KEGG, STRING, Reactome) | Provide biological context for interpreting integrated results (pathways, interactions). |
| Sample Metadata Manager (e.g., REDCap, LabKey) | Critical for ensuring accurate sample pairing across omics layers and covariate tracking. |
| Containerization Software (Docker/Singularity) | Guarantees reproducibility by encapsulating software, dependencies, and environment. |
| Normalization & Batch Correction Algorithms (e.g., ComBat, SVA) | "Wet-lab reagents" of computational biology; essential for removing technical noise before integration. |
| Benchmarking Dataset (e.g., TCGA multi-omics, simulated data) | Serves as a positive control to validate the integration pipeline and method performance. |
Detailed Methodologies and Experimental Protocols
Protocol: Multi-Omics Integrative Analysis using DIABLO (MixOmics)
Objective: To identify multi-omics biomarkers predictive of a phenotypic outcome (e.g., disease vs. control).
- Data Preprocessing: Independently normalize and filter each omics dataset (e.g., RNA-seq, Metabolomics). Scale variables to mean zero and unit variance.
- Experimental Design Check: Verify sample alignment across datasets. Format data into a list of matrices (
Xlist) and a factor vector for outcome (Y). - Parameter Tuning (
tune.block.splsda):- Perform 5-fold cross-validation to determine the optimal number of components and the number of features to select per component and per omics type.
- The tuning criterion is the balanced error rate.
- Model Training (
block.splsda): Run the DIABLO model using the tuned parameters. The model finds components that maximize covariance between selected features from all omics datasets and the outcome. - Performance Evaluation (
perf): Assess the model's prediction accuracy using repeated cross-validation to estimate generalizability. - Visualization & Interpretation: Generate sample plots (2D/3D), correlation circle plots, and loading plots to interpret the selected multi-omics features and their associations.
Protocol: Unsupervised Integration using MOFA+
Objective: To discover latent factors that capture shared and unique sources of biological variation across multiple omics assays.
- Data Preparation: Format data into a matrix per view (omics type) with matching samples. Handle missing values (MOFA+ models them explicitly).
- Model Creation (
create_mofa): Initialize the MOFA object. Specify likelihoods (Gaussian for continuous, Bernoulli for binary, Poisson for counts). - Model Training (
run_mofa):- Set training options (e.g., number of factors, convergence criteria).
- The model uses variational inference to decompose the data matrices into Factors (samples x latent factors), Weights (features x factors), and an intercept.
- Variance Decomposition Analysis (
plot_variance_explained): Quantify the proportion of variance explained per factor in each view. This identifies factors that are global (active in many views) or view-specific. - Factor Interpretation:
- Correlate factor values with known sample covariates (e.g., clinical traits).
- Examine the top-weighted features for each factor to infer biological meaning (e.g., Factor 1 loads on cell cycle genes).
- Downstream Analysis: Use factor values as reduced-dimension covariates in survival analysis, or to stratify samples into molecular subgroups.
Visualizations of Workflows and Relationships
Diagram 1: Generic Multi-Omics Integration Workflow
Diagram 2: MOFA+ Factor Model Decomposition Logic
Within the broader thesis on Introduction to multi-omics data analysis research, this case study exemplifies its translational power in oncology. Traditional single-omics approaches often fail to capture the complex, adaptive nature of cancer. Multi-omics—the integrative analysis of genomics, transcriptomics, proteomics, metabolomics, and epigenomics—provides a systems-level view of tumor biology, enabling the identification of novel, druggable targets and predictive biomarkers with higher precision.
Foundational Multi-Omics Technologies and Workflow
A standard multi-omics workflow for target identification involves sequential and parallel data generation, integration, and validation.
Experimental Protocols for Key Omics Layers:
Whole Genome/Exome Sequencing (Genomics):
- Method: DNA is extracted from tumor and matched normal tissue. Libraries are prepared, followed by sequencing on platforms like Illumina NovaSeq. Somatic variants (SNVs, indels) are called using tools like GATK Mutect2 and annotated for functional impact.
- Key Reagent: Hybridization capture probes (e.g., IDT xGen Pan-Cancer Panel) for exome/targeted sequencing enrich disease-relevant genomic regions.
RNA Sequencing (Transcriptomics):
- Method: Total RNA is extracted, ribosomal RNA is depleted, and cDNA libraries are constructed. Sequencing data is aligned (STAR), and gene expression (counts), fusion genes (Arriba, STAR-Fusion), and alternative splicing events are quantified.
Mass Spectrometry-Based Proteomics & Phosphoproteomics:
- Method: Proteins are extracted from tissue, digested with trypsin, and peptides are fractionated. Liquid chromatography-tandem MS (LC-MS/MS) is performed (e.g., on a Thermo Fisher Orbitrap Eclipse). Data is processed via MaxQuant for identification and quantification. Phosphopeptides are enriched using TiO2 or IMAC magnetic beads prior to MS.
Reverse-Phase Protein Array (RPPA - Targeted Proteomics):
- Method: Lysates are printed on nitrocellulose-coated slides, probed with validated primary antibodies against specific proteins/post-translational modifications, and detected by chemiluminescence. Provides quantitative, pathway-centric data.
Integrative Analysis: From Data to Candidate Targets
The core challenge is data integration. Methods include:
- Multi-Omics Factor Analysis (MOFA): A statistical model that identifies latent factors driving variation across all omics datasets.
- Pathway-Centric Integration: Tools like PARADIGM or Ingenuity Pathway Analysis combine omics alterations to infer pathway activity.
- Machine Learning: Supervised models (e.g., random forests) can integrate features from multiple layers to predict drug response or vulnerability.
Visualization of the Core Multi-Omics Integration Workflow:
Diagram Title: Multi-Omics Workflow for Target Discovery
Case Study: Identifying a Synthetic Lethal Target in Pancreatic Ductal Adenocarcinoma (PDAC)
A recent study integrated genomic, transcriptomic, and proteomic data from PDAC patient samples and cell lines.
Key Findings from Integrative Analysis:
- Genomics identified frequent KRAS and TP53 mutations.
- Proteomics revealed consistent overexpression of the DNA repair protein PARP1 even in tumors without homologous recombination (HR) genomic signatures.
- Phosphoproteomics identified hyperactivation of the ATM/ATR DNA damage response (DDR) pathway.
Hypothesis: PDAC cells with KRAS/TP53 co-mutations exhibit a latent DNA repair defect and rely on PARP1-mediated backup repair, creating a context-specific vulnerability.
Visualization of the Identified Signaling Axis:
Diagram Title: PDAC Synthetic Lethality Hypothesis
Validation Protocol:
- Genetic Knockdown: siRNA-mediated PARP1 knockdown in PDAC cell lines (with KRAS/TP53 mutations) led to significant loss of viability vs. controls.
- Pharmacological Inhibition: Treatment with PARP inhibitors (Olaparib, Talazoparib) selectively killed PDAC cells, correlating with proteomic PARP1 levels, not genomic HR status.
- In Vivo Validation: Patient-derived xenograft (PDX) models with high proteomic PARP1 showed marked tumor regression on PARPi treatment, confirming it as a novel, actionable target beyond BRCA-mutant contexts.
Table 1: Multi-Omics Data Yield from PDAC Cohort (n=50)
| Omics Layer | Platform | Key Metrics | Median Coverage/Depth |
|---|---|---|---|
| Genomics | WES (Illumina) | 12,500 somatic variants; 45% KRAS mut; 60% TP53 mut | 150x tumor, 60x normal |
| Transcriptomics | RNA-Seq (Poly-A) | 18,000 genes expressed; 5,000 differentially expressed | 50M paired-end reads |
| Proteomics | LC-MS/MS (TMT) | 8,500 proteins quantified; PARP1 >2x overexpressed in 70% | N/A |
| Phosphoproteomics | LC-MS/MS (TiO2) | 25,000 phosphosites; DDR pathway enriched (p<0.001) | N/A |
Table 2: Validation Experiment Results
| Experiment | Model System | Intervention | Key Result (vs Control) | p-value |
|---|---|---|---|---|
| PARP1 Knockdown | MIA PaCa-2 Cell Line | siRNA PARP1 | 75% reduction in viability | < 0.001 |
| PARP Inhibition | 10 PDAC Cell Lines | Olaparib (10µM, 72h) | IC50 correlated with PARP1 protein (R=0.82) | 0.003 |
| In Vivo PDX Study | 5 PARP1-High PDX Models | Talazoparib (1mg/kg, 21d) | 80% tumor growth inhibition | < 0.001 |
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Reagents for Multi-Omics Target Discovery
| Reagent/Solution | Vendor Examples | Primary Function in Workflow |
|---|---|---|
| AllPrep DNA/RNA/Protein Kit | Qiagen | Simultaneous isolation of intact multi-omic molecules from a single tissue sample. |
| xGen Pan-Cancer Hybridization Panel | Integrated DNA Technologies (IDT) | For targeted exome sequencing, enriching cancer-related genes for efficient variant detection. |
| Poly(A) mRNA Magnetic Beads | NEB, Thermo Fisher | Isolation of polyadenylated mRNA from total RNA for RNA-Seq library prep. |
| TMTpro 16plex Isobaric Label Reagent Set | Thermo Fisher | Multiplexing up to 16 samples in one MS run for high-throughput, quantitative proteomics. |
| Phosphopeptide Enrichment TiO2 Magnetic Beads | GL Sciences, MilliporeSigma | Selective enrichment of phosphopeptides from complex peptide mixtures for phosphoproteomics. |
| Validated Primary Antibodies for RPPA/WB | CST, Abcam | Target-specific protein detection and quantification for orthogonal validation. |
| PARP Inhibitors (Olaparib, Talazoparib) | Selleckchem, MedChemExpress | Pharmacological probes for validating PARP1 target dependency in in vitro and in vivo assays. |
This case study demonstrates that multi-omics integration moves beyond correlative genomics to reveal functional, context-dependent drug targets. The identification of PARP1 as a target in a molecularly defined PDAC subset, driven by proteomic rather than genomic alterations, underscores the necessity of layered data. This approach, framed within systematic multi-omics research, is reshaping oncology drug discovery by identifying novel targets, defining responsive patient populations, and accelerating the development of precision therapies.
Solving the Multi-Omics Puzzle: Troubleshooting Batch Effects, Statistical Power, and Integration Challenges
Identifying and Correcting for Batch Effects and Technical Variation Across Platforms
Within the broader thesis on Introduction to multi-omics data analysis research, a fundamental challenge emerges when integrating datasets generated across different laboratories, times, or technological platforms. This challenge is the introduction of non-biological, systematic technical variation, commonly termed "batch effects." These artifacts can be of greater magnitude than the biological signals of interest, leading to spurious findings, reduced statistical power, and irreproducible results. This guide provides an in-depth technical examination of methodologies for identifying, diagnosing, and correcting for these pervasive variations.
Batch effects arise from a multitude of sources, which vary by platform.
Primary Sources of Variation:
- Platform/Technology: Differences between microarray vs. RNA-seq, LC-MS vs. GC-MS, or different instrument manufacturers.
- Reagent Lots: Variation in antibody lots, sequencing kits, or chromatography columns.
- Operator & Protocol: Differences in sample handling, library preparation, and data acquisition personnel.
- Temporal Runs: Experiments processed on different days or in different sequential orders.
Diagnosis is the critical first step. Principal Component Analysis (PCA) and hierarchical clustering are standard exploratory tools, where samples frequently cluster by batch rather than biological condition. Formal statistical tests like the Surrogate Variable Analysis (SVA) or the Percent Variance Explained (PVE) calculation can quantify the proportion of variance attributable to batch.
Table 1: Percent Variance Explained by Batch in Example Multi-Omics Datasets
| Omics Type | Platform A | Platform B | PVE by Batch (%) | Statistical Test Used |
|---|---|---|---|---|
| Transcriptomics | Illumina HiSeq | Illumina NovaSeq | 35% | SVA (Leek, 2014) |
| Proteomics | Thermo TMT-10plex | Bruker label-free | 50% | ANOVA-PVE |
| Metabolomics | Agilent GC-TOFMS | Waters LC-HRMS | 28% | PCA-based PVE |
| Methylomics | Illumina 450K | Illumina EPIC | 22% | Combat (Johnson, 2007) |
Correction Methodologies and Experimental Protocols
Correction strategies are divided into study design-based and computational approaches.
Study Design Best Practices
- Randomization: Process samples from all biological groups in each batch.
- Balancing: Ensure equal representation of conditions within each batch.
- Reference/Control Samples: Include identical technical control samples (e.g., pooled reference) across all batches/platforms for calibration.
Core Computational Correction Protocols
Protocol A: ComBat and its Derivatives (Empirical Bayes Framework)
- Input: A normalized, but uncorrected, data matrix (e.g., gene expression counts).
- Model Specification: Define the model matrix for biological covariates of interest (e.g., disease state).
- Batch Parameterization: Specify the batch covariate (e.g., sequencing run).
- Empirical Bayes Adjustment: The algorithm estimates batch-specific location (mean) and scale (variance) parameters and shrinks them toward the global mean/variance.
- Output: A batch-adjusted matrix where data distributions are aligned across batches. Note: ComBat-Seq is specifically designed for count-based data (e.g., RNA-seq), preserving the integer property.
Protocol B: Remove Unwanted Variation (RUV) Series
- Input: Data matrix and specification of negative control features or replicate samples.
- Control Feature Identification: Utilize housekeeping genes (RUVg), replicate samples (RUVs), or factors derived from residuals (RUVr) as estimates of unwanted variation.
- Factor Estimation: Perform factor analysis (e.g., SVD) on the control data to estimate
kunwanted factors. - Regression: Regress out the
kunwanted factors from the original data matrix using a linear model. - Output: Residuals representing batch-corrected data.
Protocol C: Harmony for High-Dimensional Integration
- Input: A PCA or other embedding of the original data (e.g., top PCs from scRNA-seq).
- Clustering: Cells/samples are soft-clustered based on their embeddings.
- Correction: For each cluster, centroid positions are calculated per batch and iteratively corrected via maximum diversity clustering to remove batch-specific centroids.
- Output: A corrected, integrated low-dimensional embedding.
Experimental Workflow for Cross-Platform Integration
The following diagram illustrates the standard workflow for diagnosing and correcting batch effects in a multi-platform study.
Diagram Title: Multi-Omics Batch Effect Correction Workflow
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagents and Tools for Batch Effect Management
| Item Name | Function & Purpose | Example Product/Software |
|---|---|---|
| Reference RNA/DNA | A universal, stable biological control processed in every batch to calibrate and monitor technical performance. | Universal Human Reference RNA (Agilent), NA12878 genomic DNA. |
| Internal Standard Spike-Ins | Known quantities of exogenous molecules (e.g., ERCC RNA, heavy-labeled peptides) added to each sample for normalization across runs. | ERCC RNA Spike-In Mix (Thermo), Proteomics Dynamic Range Standard (Sigma). |
| Multiplexing Kits | Chemical tags to label and pool multiple samples for simultaneous processing in a single run, eliminating run-to-run variation. | Tandem Mass Tag (TMT) kits, Multiplexed siRNA kits. |
| ComBat | Empirical Bayes software for batch effect correction in genomics/proteomics data. | sva R package (ComBat function). |
| Harmony | Algorithm for integrating single-cell or high-dimensional data across batches. | harmony R/Python package. |
| limma (removeBatchEffect) | Linear modeling approach to adjust for batch effects while preserving biological variables. | limma R package. |
| RUVcorr | Suite of methods using control genes/replicates to remove unwanted variation. | ruv R package. |
Advanced Considerations and Current Trends
- Non-Linear and Deep Learning Methods: Tools like SCANVI and scGen use variational autoencoders to learn and correct for complex, non-linear batch effects in single-cell data.
- Multi-Modal Integration: Methods like TotalVI (for CITE-seq) and MOFA+ are designed to integrate multiple omics modalities while accounting for technical noise from each platform.
- Benchmarking: Systematic benchmarks (e.g., by the IBDLab) consistently show that the optimal method depends on the data type, batch effect strength, and biological context. No single method is universally superior.
Conclusion: For robust and reproducible multi-omics research, proactive study design to minimize batch effects, coupled with rigorous post-hoc diagnosis and application of validated correction algorithms, is non-negotiable. The choice of correction tool must be guided by the data structure and followed by thorough validation to ensure biological signals are not distorted.
Addressing Missing Data and Imputation in Sparse Omics Datasets
1. Introduction
Within the framework of multi-omics data analysis research, integrating datasets from genomics, transcriptomics, proteomics, and metabolomics presents a fundamental challenge: pervasive missing data. This sparsity arises from technical limitations (e.g., detection thresholds in mass spectrometry), biological abundance below instrument sensitivity, and data processing artifacts. The pattern and mechanism of missingness—Missing Completely At Random (MCAR), Missing At Random (MAR), or Missing Not At Random (MNAR)—critically influence the selection and performance of imputation methods. Unaddressed, missing values cripple downstream statistical power and integrative modeling, leading to biased biological inferences. This technical guide details contemporary strategies for diagnosing, managing, and imputing missing values in sparse omics datasets.
2. Mechanisms and Patterns of Missingness
Accurate characterization of missing data is the first essential step. The following table summarizes the types, causes, and diagnostic indicators.
Table 1: Mechanisms of Missing Data in Omics
| Mechanism | Acronym | Definition | Common Cause in Omics | Diagnostic Test (Example) |
|---|---|---|---|---|
| Missing Completely At Random | MCAR | Missingness is independent of observed and unobserved data. | Random technical failures, sample loss. | Little's MCAR test; no pattern in missing data matrix (MNAR). |
| Missing At Random | MAR | Missingness depends only on observed data. | Low abundance ions masked by high abundance ones in LC-MS. | Pattern in MNAR correlated with feature intensity or sample group. |
| Missing Not At Random | MNAR | Missingness depends on the unobserved missing value itself. | Signal below instrument detection limit (censored data). | Statistical tests for left-censoring; association with detection limits. |
3. Experimental Protocols for Evaluating Imputation Performance
To benchmark imputation algorithms, a robust experimental protocol is required.
Protocol 1: Imputation Benchmarking via Simulation
- Start with a Complete Dataset: Identify or create a high-quality, dense omics matrix (e.g., proteomics data with minimal missingness).
- Induce Missing Data: Artificially introduce missing values under controlled mechanisms:
- MCAR: Randomly remove values across the matrix (e.g., 5%, 10%, 20%).
- MAR: Remove values with a probability based on observed row/column means.
- MNAR (Left-censoring): Remove values below a simulated intensity threshold.
- Apply Imputation Methods: Run the sparsified matrix through multiple imputation algorithms (see Section 4).
- Evaluate Performance: Compare imputed values against the held-out true values using metrics: Root Mean Square Error (RMSE), Pearson correlation, and preservation of biological variance (PCA distortion).
Protocol 2: Downstream Analysis Validation
- Impute Multiple Versions: Generate complete datasets using different imputation methods for a real, sparse dataset.
- Perform Differential Analysis: Apply the same statistical test (e.g., limma for transcriptomics) to each imputed dataset.
- Compare Results: Evaluate concordance in the list of significant features (e.g., genes, proteins) and their p-value distributions across methods. A stable method yields robust, replicable signatures.
4. Imputation Methodologies and Workflow
A strategic workflow guides the choice of imputation method based on data type and missingness mechanism.
Decision Workflow for Imputation Method Selection
Table 2: Comparison of Common Imputation Methods for Omics Data
| Method Category | Example Algorithms | Principle | Best For | Advantages | Limitations |
|---|---|---|---|---|---|
| Simple Replacement | Min Value, Mean/Median | Replaces missing values with a constant derived from the observed data. | Quick assessment, MNAR (Min). | Fast, simple. | Distorts distribution, underestimates variance. |
| Local Similarity | k-Nearest Neighbors (KNN), MissForest | Uses similar rows/columns (features/samples) to estimate missing values. | MCAR, MAR, low-to-moderate sparsity. | Utilizes data structure, non-parametric. | Computationally heavy, sensitive to distance metrics. |
| Matrix Factorization | Singular Value Decomposition (SVD), MICE | Decomposes matrix into lower-rank approximations to predict missing entries. | MAR, high sparsity, large datasets. | Captures global patterns, robust. | Assumptions of linearity (SVD), convergence issues (MICE). |
| MNAR-Specific | QRILC, Downshifted Normal (DL) | Models the missing data as censored from a known distribution (e.g., log-normal). | MNAR (left-censored), proteomics/metabolomics. | Biologically plausible for detection limits. | Distribution assumptions may not hold. |
| Deep Learning | Denoising Autoencoder (DAE), GAN | Neural networks learn a robust data model to reconstruct missing entries. | All types, very high-dimensional data. | Highly flexible, captures complex patterns. | "Black box", requires large data and tuning. |
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Missing Data Analysis
| Tool/Reagent | Function/Benefit | Example/Note |
|---|---|---|
R missMDA / mice packages |
Comprehensive suite for diagnosis, imputation (PCA, MICE), and evaluation of missing data. | Essential for statistical rigor and multiple imputation workflows. |
Python scikit-learn / fancyimpute |
Provides KNN, matrix factorization, and deep learning-based imputation algorithms. | Integrates with Python-based omics pipelines (scanpy, SciPy). |
Proteomics-specific: NAguideR |
Web tool & R package evaluating >10 imputation methods tailored for proteomics MNAR/MAR data. | Critical for LC-MS data; provides performance metrics. |
Metabolomics-specific: MetImp |
Online tool for diagnosing missingness mechanism and applying metabolomics-optimized imputation. | Handles MNAR via probabilistic models. |
| Simulation Data (Benchmark) | A complete, real omics dataset with known values, used to induce missingness and test algorithms. | e.g., "PXD001481" proteomics dataset from PRIDE repository. |
| High-Performance Computing (HPC) Cluster | Cloud or local cluster resources for computationally intensive methods (MissForest, DAE). | Necessary for large-scale multi-omics integration projects. |
Ensuring Sufficient Sample Size and Statistical Power for Integrated Analysis
A fundamental thesis in modern biomedical research is that integrating multiple molecular data layers—genomics, transcriptomics, proteomics, metabolomics—provides a more comprehensive systems-level understanding of biological processes and disease etiology than any single modality alone. However, the high-dimensionality, heterogeneity, and technical noise inherent in each omics layer present significant statistical challenges. The most critical, yet often overlooked, prerequisite for robust integrated multi-omics analysis is the careful a priori determination of sufficient sample size and statistical power. Underpowered studies lead to high false discovery rates, irreproducible results, and wasted resources, fundamentally undermining the translational promise of multi-omics.
Core Statistical Concepts & Challenges
Statistical Power is the probability that a test will correctly reject a false null hypothesis (i.e., detect a true effect). In multi-omics integration, "effect" may refer to a true association between a molecular feature and a phenotype, or a true correlation between features across omics layers.
Key challenges include:
- High-Dimensionality (p >> n): The number of features (p) vastly exceeds the number of samples (n), increasing the risk of overfitting and multiplicity.
- Data Heterogeneity: Different data types (e.g., continuous RNA-seq counts, discrete SNP genotypes) require different statistical models.
- Complex Integration Models: Methods like Multi-Omic Factor Analysis (MOFA) or sparse Partial Least Squares Discriminant Analysis (sPLS-DA) have complex power characteristics that are not captured by simple formulas.
- Multiple Testing Burden: Testing thousands of features necessitates severe correction (e.g., Bonferroni, FDR), which dramatically increases the sample size needed to maintain power.
Quantitative Data on Power and Sample Size
The required sample size is influenced by effect size, desired power, significance threshold, and data structure. The table below summarizes generalized estimates for different primary analysis goals in multi-omics studies.
Table 1: Generalized Sample Size Requirements for Common Multi-Omics Analysis Goals
| Primary Analysis Goal | Typical Minimum Sample Size Range (Per Group) | Key Determining Factors | Typical Achievable Effect Size (Cohen's d / AUC) | ||
|---|---|---|---|---|---|
| Differential Abundance (Single Omics) | 15 - 50 | Expected fold-change, biological variance, false discovery rate. | d = 0.8 - 1.5 (Moderate-Large) | ||
| Multi-Omics Class Prediction | 50 - 150 | Number of omics layers, classifier complexity, expected prediction accuracy. | AUC > 0.75 - 0.85 | ||
| Network/Pairwise Integration | 100 - 300 | Sparsity of true correlations, noise level, desired stability. | r | > 0.3 - 0.5 | |
| Unsupervised Clustering (Subtyping) | 50 - 200 | Separation between clusters, proportion of informative features. | Silhouette Width > 0.25 |
Table 2: Impact of Multiple Testing Correction on Required Sample Size (Example: Differential Expression)
| Number of Features Tested (m) | Uncorrected α | Bonferroni α' (α/m) | Required N per group to detect effect size d=0.8 at 80% power |
|---|---|---|---|
| 100 | 0.05 | 0.0005 | ~52 |
| 10,000 | 0.05 | 5e-06 | ~78 |
| 50,000 | 0.05 | 1e-06 | ~85 |
Experimental Protocols for Power Assessment
Protocol 4.1: Simulation-Based Power Analysis for Integrated Analysis
This is the gold-standard method for complex multi-omics study designs.
- Define a Data-Generating Model: Use a realistic model (e.g., multivariate normal, Poisson-Gamma for counts) that reflects the covariance structure between omics features from pilot or public data.
- Incorporate Known Effects: Introduce true effects (e.g., differential expression for 5% of features, cross-omics correlations for a defined subset) of a hypothesized magnitude.
- Simulate Datasets: Repeatedly (e.g., 1000 times) simulate full multi-omics datasets for a range of candidate sample sizes (e.g., N=20, 40, 60...).
- Apply Analysis Pipeline: For each simulated dataset, run the planned integration analysis (e.g., sPLS-DA, MOFA, association testing).
- Calculate Empirical Power: For each sample size, power = (Number of simulations where true effect is correctly detected) / (Total simulations).
Protocol 4.2: Pilot Study & Resampling-Based Estimation
When simulations are infeasible due to unknown parameters.
- Acquire Pilot Data: Obtain data from a small cohort (e.g., n=10-15 per group) or a relevant public dataset.
- Bootstrap Resampling: Randomly draw subsamples of varying sizes (with replacement) from the pilot data.
- Perturbation: Add synthetic effects of a specific size to the subsampled data to mimic a true signal.
- Stability Assessment: Measure the stability of results (e.g., overlap in selected features, consistency of cluster assignments) across bootstrap iterations for each subsample size.
- Extrapolate: Identify the sample size at which result stability (a proxy for reproducibility and power) plateaus or reaches an acceptable threshold (e.g., Jaccard index > 0.8 for feature selection).
Visualization of Workflows and Relationships
Title: Multi-Omics Sample Size Determination Workflow
Title: Key Factors Determining Statistical Power
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Multi-Omics Study Design and Power Analysis
| Tool / Reagent Category | Specific Example(s) | Primary Function in Power/Sample Size Context |
|---|---|---|
| Statistical Software Packages | R (pwr, sizepower, SIMLR), Python (statsmodels, scikit-learn), G*Power |
Provide functions for standard power calculations and enable custom simulation studies. |
| Multi-Omics Simulation Frameworks | SPsimSeq, POWSC, MosiSim, combiROC |
Generate realistic, synthetic multi-omics datasets with known ground truth for power evaluation. |
| Bioinformatics Data Repositories | TCGA, GEO, EBI Metabolights, PRIDE | Source of pilot/public data for parameter estimation and resampling-based power analysis. |
| High-Dimensional Integrative Analysis Tools | MOFA+, mixOmics, iClusterBayes, OmicsPLS | The planned endpoint analysis tools whose performance is being evaluated for power. |
| Cloud Computing Credits | AWS, Google Cloud, Azure Credits | Provide the computational resources necessary for large-scale, repeated simulations. |
| Standardized Reference Materials | NIST SRM 1950 (Metabolites), HEK293 or Pooled Human Plasma samples | Used in pilot studies to accurately estimate technical variance, a key component of "noise." |
Within the burgeoning field of multi-omics data analysis research, integrating genomics, transcriptomics, proteomics, and metabolomics datasets presents unprecedented opportunities for discovery. However, this high-dimensional data landscape, where the number of features (p) vastly exceeds the number of samples (n), is a fertile ground for statistical overfitting. Overfitting occurs when a model learns not only the underlying signal but also the noise and idiosyncrasies specific to the training dataset, leading to impressive performance during discovery that fails to generalize upon independent validation. This guide provides an in-depth technical framework for balancing discovery-driven hypothesis generation with rigorous validation to build robust, translatable models in multi-omics research.
The Core Challenge: Overfitting in High-Dimensional Spaces
Overfitting is intrinsically linked to model complexity and the curse of dimensionality. In a p >> n scenario, simple models can perfectly fit the training data by chance, identifying spurious correlations.
Table 1: Common Consequences of Overfitting in Multi-Omics Analysis
| Consequence | Description | Typical Manifestation |
|---|---|---|
| Inflated Performance Metrics | Training/Test accuracy or AUC is artificially high. | AUC of 0.99 in discovery cohort drops to 0.65 in validation. |
| Non-Replicable Feature Signatures | Identified biomarkers or gene signatures fail in independent cohorts. | A 50-gene prognostic panel from transcriptomics shows no significant survival association upon validation. |
| Reduced Predictive Power | Model fails to predict outcomes for new samples. | A drug response classifier performs at chance level in a new clinical trial population. |
| Over-Interpretation of Noise | Biological narratives are built on statistically insignificant patterns. | A pathway is falsely implicated in disease mechanism. |
Foundational Methodologies for Robust Analysis
Experimental Design & Cohort Splitting
The first line of defense is a sound experimental design that pre-defines validation cohorts.
Protocol: Rigorous Train-Validation-Test Split for Multi-Omics Studies
- Cohort Assembly: Collect data from all available samples (N total).
- Stratified Splitting: Partition data into three mutually exclusive sets before any analysis or feature selection.
- Discovery/Training Set (60-70%): Used for model development, feature selection, and initial parameter tuning.
- Validation/Held-Out Set (15-20%): Used to assess model performance during development, guide model selection, and prevent overfitting during tuning.
- Test Set (15-20%): Used only once for a final, unbiased evaluation of the fully specified model. It must never influence the discovery process.
- Stratification: Ensure key clinical variables (e.g., disease status, treatment arm) are proportionally represented across splits to avoid bias.
- Lock the Test Set: The test set should be physically or digitally sequestered until the final evaluation phase.
Regularization Techniques
Regularization penalizes model complexity to prevent over-reliance on any single feature.
Protocol: Implementing Regularized Regression (LASSO)
- Data Preparation: Standardize all omics features (mean=0, variance=1) to ensure penalty is applied equally.
- Model Specification: Fit a logistic (for classification) or Cox (for survival) regression with an L1-norm (LASSO) penalty:
argmin( Loss(Data|β) + λ * Σ|βj| ). The tuning parameterλcontrols penalty strength. - Cross-Validation: Use k-fold (e.g., k=10) cross-validation within the training set to find the optimal
λthat minimizes cross-validated prediction error. - Model Fitting: Refit the model on the entire training set using the optimal
λ. Features with coefficients shrunk to zero are effectively selected out. - Evaluation: Apply the fitted model (with the selected features and their non-zero coefficients) to the validation/test set.
Dimensionality Reduction & Feature Selection
Reducing the feature space is critical. Methods vary in how they handle correlation and noise.
Table 2: Comparison of Dimensionality Reduction Techniques
| Method | Type | Key Principle | Strength | Weakness for Overfitting |
|---|---|---|---|---|
| Principal Component Analysis (PCA) | Unsupervised | Finds orthogonal axes of maximum variance. | De-noising, handles collinearity. | Components may not be biologically interpretable or relevant to outcome. |
| Partial Least Squares (PLS) | Supervised | Finds components explaining covariance between X and Y. | Captures outcome-relevant signal. | Risk of fitting noise if not properly cross-validated. |
| Recursive Feature Elimination (RFE) | Supervised | Iteratively removes least important features. | Directly selects a relevant feature set. | High computational cost; requires nested CV to be reliable. |
| Variance Filtering | Unsupervised | Removes low-variance features. | Simple, fast pre-filter. | May discard biologically important low-variance signals. |
Protocol: Nested Cross-Validation for Unbiased Error Estimation This protocol is essential when performing both feature selection and model tuning.
- Define Outer Loop: Split the full dataset (excluding the final locked test set) into K outer folds (e.g., K=5).
- Iterate Outer Folds: For each outer fold i:
a. Hold out fold i as the validation set.
b. Use the remaining K-1 folds as the development set.
c. Inner Loop: Perform feature selection and hyperparameter tuning (e.g., choosing
λfor LASSO) using only the development set, with another layer of cross-validation (the inner CV). d. Train the final model with the chosen features/parameters on the entire development set. e. Evaluate this model on the held-out outer validation fold i. - Aggregate Results: The performance metrics across the K outer folds provide an almost unbiased estimate of generalization error.
Validation Frameworks
Validation confirms that discovered patterns are generalizable.
Protocol: External Validation in a Multi-Center Study
- Discovery Cohort: Perform full analysis (feature selection, model building) on data from Center A.
- Model Freezing: Finalize the model (exact features, algorithm, coefficients/thresholds). No further adjustments are allowed.
- Blinded Application: Apply the frozen model to the raw omics data from Center B.
- Performance Assessment: Calculate the same performance metrics (accuracy, AUC, hazard ratio) on the Center B data.
- Statistical Comparison: Use DeLong's test (for AUC) or log-likelihood ratio tests to assess if the performance drop from discovery to validation is statistically significant.
Visualization of Core Concepts
Diagram 1: The Overfitting Risk and Mitigation Pathway in Omics
Diagram 2: Nested Cross-Validation Workflow for Unbiased Error
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Tools for Robust Multi-Omics Analysis
| Item | Category | Function in Avoiding Overfitting |
|---|---|---|
| Independent Validation Cohort | Biological Sample Set | Provides unbiased biological material to test generalizability of discovered signatures. |
| Locked Test Set | Data Management Protocol | A portion of data sequestered for final evaluation only, preventing data leakage and giving a true performance estimate. |
| scikit-learn (Python) | Software Library | Provides standardized, peer-reviewed implementations of CV splitters (StratifiedKFold), regularized models (LASSO, ElasticNet), and feature selection tools. |
| caret / tidymodels (R) | Software Framework | Offers a unified interface for performing complex modeling workflows with built-in resampling and validation in R. |
| ComBat / SVA | Bioinformatics Tool | Corrects for batch effects across different experimental runs or cohorts, ensuring technical noise isn't modeled as biological signal. |
| Permutation Testing Framework | Statistical Method | Generates null distributions by randomly shuffling labels to assess the statistical significance of model performance, guarding against lucky splits. |
| Pre-registration Protocol | Research Practice | Publicly documenting analysis plans before seeing the data minimizes "fishing expeditions" and p-hacking. |
In multi-omics data analysis research, the path from high-dimensional discovery to validated knowledge is fraught with statistical pitfalls. Balancing discovery and validation is not a secondary step but the core imperative. By mandating rigorous experimental design (train-validation-test splits), employing regularization and dimensionality reduction with nested cross-validation, and insisting on external validation, researchers can build models that not only fit their data but truly explain the underlying biology. This disciplined approach is essential for generating reliable biomarkers, therapeutic targets, and insights that can successfully transition from the research bench to clinical impact.
Best Practices for Computational Resource Management and Workflow Reproducibility
Within the rapidly evolving field of multi-omics data analysis—integrating genomics, transcriptomics, proteomics, and metabolomics—the scale and complexity of computations present significant challenges. Effective management of computational resources and ensuring the reproducibility of intricate workflows are not merely operational concerns but fundamental pillars of rigorous, scalable, and collaborative scientific research in drug development and systems biology. This guide outlines current best practices to address these critical needs.
Computational Resource Management
Efficient management of hardware and software resources is essential for handling large multi-omics datasets, which can easily reach petabyte scales in population-level studies.
Resource Allocation and Monitoring
Key metrics must be tracked to optimize resource utilization and identify bottlenecks. The following table summarizes critical quantitative benchmarks for a typical multi-omics analysis node.
Table 1: Computational Resource Benchmarks for Multi-Omics Analysis
| Resource Type | Recommended Baseline (2024) | High-Performance Target | Monitoring Tool Example | Key Metric to Track |
|---|---|---|---|---|
| CPU Cores per Node | 16-32 cores | 64-128+ cores | htop, Slurm |
% CPU utilization per process |
| RAM | 64-128 GB | 512 GB - 2 TB | free, Prometheus |
Peak memory footprint |
| Storage (Fast) | 1-5 TB NVMe SSD | 10-50 TB NVMe SSD | iostat, Grafana |
I/O wait times, read/write speed |
| Storage (Archive) | 100 TB+ (Object/GlusterFS) | 1 PB+ (Lustre/Ceph) | Vendor dashboards | Cost per TB, retrieval latency |
| Cloud/Cluster Scheduler | Slurm, Kubernetes | Kubernetes with auto-scaling | Built-in dashboards | Job queue time, cost per analysis |
Containerization for Environment Consistency
Containerization encapsulates software dependencies, ensuring identical environments across development, testing, and high-performance computing (HPC) deployment.
Experimental Protocol: Creating a Reproducible Container for RNA-Seq Analysis
- Define Dependencies: Create a
requirements.txt(for Python) and/or aBiocondaenvironment file (environment.yml) listing all packages (e.g., STAR, DESeq2, MultiQC) with exact versions. - Write a Dockerfile: Use a minimal base image (e.g.,
rocker/r-ver:4.3.1). Copy dependency files, install tools via package managers (apt, conda), and set the working directory. - Build Image: Execute
docker build -t rnaseq-pipeline:2024.06 . - Test Locally: Run analysis on a small test dataset:
docker run -v $(pwd)/data:/data rnaseq-pipeline:2024.06 python /scripts/run_analysis.py - Push to Registry: Upload the verified image to a repository like Docker Hub or Google Container Registry for team access.
- Deploy on HPC/Cloud: Use Singularity/Apptainer (common on HPC) to pull and run the container:
singularity exec docker://registry/rnaseq-pipeline:2024.06 python script.py.
Workflow Reproducibility
Reproducibility requires capturing the complete data lifecycle: from raw data, through code and parameters, to the final results.
Workflow Management Systems
Scripted pipelines ensure explicit, version-controlled execution paths.
Table 2: Comparison of Workflow Management Systems
| System | Primary Language | Strengths | Ideal Use Case in Multi-Omics |
|---|---|---|---|
| Nextflow | DSL (Groovy-based) | Strong HPC/Cloud support, built-in conda/docker | Large-scale, portable omics pipelines (nf-core) |
| Snakemake | Python (YAML-like) | Readable syntax, excellent Python integration | Complex, multi-step integrative analyses |
| CWL (Common Workflow Language) | YAML/JSON | Platform-agnostic standard, excellent for tool wrapping | Sharing tools across institutions |
| WDL | Human-readable syntax | Cloud-native, used by Terra/Broad Institute | Large cohort analysis on cloud platforms |
Experimental Protocol: Implementing a Snakemake Pipeline for Proteomics/Transcriptomics Integration
- Define Rule Graph: Map input/output relationships for each step: raw proteomics (mass spec) → identification (MaxQuant) → raw transcriptomics (fastq) → alignment (STAR) → differential analysis (limma/DESeq2) → integrative analysis (MixOmics).
- Write Snakefile:
- Execute with Reproducibility Flags: Run
snakemake --use-conda --use-singularity --cores 32to automatically manage software and container environments. - Archive: Snakemake can automatically log software versions and package the entire workflow.
Data and Provenance Tracking
Persistent identifiers (DOIs) for datasets and code (via Zenodo, Figshare) are mandatory. Computational provenance—the detailed record of all operations applied to data—should be captured automatically using tools like renv (for R), Poetry (for Python), or workflow system reports.
Diagram 1: Workflow reproducibility data lifecycle.
The Scientist's Toolkit: Research Reagent Solutions for Computational Multi-Omics
Table 3: Essential Computational Tools & Platforms
| Item | Category | Function & Explanation |
|---|---|---|
| Conda/Bioconda/Mamba | Package Manager | Installs and manages versions of bioinformatics software and libraries in isolated environments, resolving dependency conflicts. |
| Docker/Singularity | Containerization | Packages an entire analysis environment (OS, tools, libraries) into a portable, reproducible unit. Singularity is security-aware for HPC. |
| Git & GitHub/GitLab | Version Control | Tracks all changes to analysis code, configuration files, and documentation, enabling collaboration and rollback. |
| Nextflow / Snakemake | Workflow Manager | Defines, executes, and manages complex, multi-step computational pipelines, ensuring portability and scalability. |
| Jupyter / RStudio | Interactive Development Environment (IDE) | Provides an interactive interface for exploratory data analysis, visualization, and literate programming (notebooks). |
| Terra / Seven Bridges | Cloud Platform | Integrated cloud environments providing data, tools, workflows, and scalable compute for collaborative multi-omics projects. |
| FastDUR / md5sum | Data Integrity Tool | Generates checksums to verify that data files have not been corrupted during transfer or storage. |
Integrated Best Practice Protocol
The following workflow synthesizes the principles outlined above for a reproducible, resource-aware multi-omics study.
Experimental Protocol: End-to-End Reproducible Multi-Omics Analysis Project
- Project Initiation:
- Create a canonical project directory structure (
data/raw,data/processed,code,results,docs). - Initialize a Git repository and a
README.mdwith the study abstract and setup instructions. - Document all computational resource requests (cores, memory, storage) based on pilot data.
- Create a canonical project directory structure (
- Environment Setup:
- Create a
environment.ymlfile specifying all Conda packages. - Write a
Dockerfilethat builds atop this Conda environment and installs any non-Conda tools. - Build and tag the Docker image, push to a team-accessible registry.
- Create a
- Pipeline Development:
- Implement the analysis as a Nextflow or Snakemake pipeline, with each process/task specifying its container image and resource requirements (cpus, memory).
- Use configuration profiles (
nextflow.config) to define settings for local, cluster, or cloud execution. - Process all data through this pipeline; never perform manual, unrecorded steps on data.
- Execution and Monitoring:
- Launch the pipeline on the target infrastructure (HPC scheduler, Kubernetes).
- Use monitoring tools (e.g., Slurm
sacct, Prometheus/Grafana for cloud) to track resource use against estimates and optimize future runs.
- Provenance Capture and Publication:
- Use the workflow system's reporting feature (
nextflow log,snakemake --report) to generate an execution report. - Archive the final dataset, code snapshot, and container image in a repository like Zenodo to receive a DOI.
- Publish the workflow on a platform like nf-core for community use and peer review.
- Use the workflow system's reporting feature (
Diagram 2: Integrated best practice workflow for reproducible analysis.
Benchmarking Multi-Omics Findings: Validation Strategies and Comparative Tool Analysis for Robust Results
Within the framework of multi-omics data analysis research, the identification of robust biomarkers, therapeutic targets, or key regulatory networks is a primary goal. High-throughput technologies (e.g., RNA-seq, proteomics) generate vast datasets with inherent technical and biological noise. Consequently, findings from a single omics platform or a single patient cohort are prone to false positives and lack translational confidence. Orthogonal validation—the practice of confirming a result using independent methodological and sample-based approaches—is a critical, non-negotiable step. This guide details the strategic implementation of orthogonal validation using independent cohorts and foundational molecular biology assays, thereby bridging discovery-phase multi-omics analytics with verifiable biological reality.
Strategic Framework for Orthogonal Validation
A robust orthogonal validation plan operates on two axes:
- Axis 1: Sample Independence: Validation in a biologically independent cohort not used in the initial discovery analysis.
- Axis 2: Methodological Independence: Validation using an experimental technique based on different physicochemical principles than the discovery platform.
Table 1: Orthogonal Validation Matrix for Multi-Omics Findings
| Discovery Omics Platform | Primary Finding Example | Methodologically Orthogonal Assay | Sample Orthogonacy Requirement |
|---|---|---|---|
| RNA-seq | Differential gene expression (mRNA) | qPCR (for transcripts) / Western Blot (for protein) | Use an independent patient cohort or a separate in vitro/in vivo model system. |
| Shotgun Proteomics | Up-regulated protein X | Western Blot or Targeted MRM/SRM-MS | Validate in a cohort from a different clinical site or a distinct cell line panel. |
| Phospho-proteomics | Increased phosphorylation at site Y | Phospho-specific Western Blot or Immunofluorescence | Confirm in an independent set of stimulated vs. control samples. |
| Metabolomics (LC-MS) | Elevated metabolite Z | Enzymatic Assay or Targeted MS | Validate in a separate biological replicate set or patient plasma cohort. |
Detailed Experimental Protocols for Key Assays
Quantitative Reverse Transcription PCR (qPCR)
Purpose: To absolutely quantify the expression levels of specific mRNA transcripts identified from RNA-seq data. Detailed Protocol:
- RNA Isolation & QC: Extract total RNA from validation cohort samples using a silica-membrane column kit. Assess purity (A260/A280 ~1.9-2.1) and integrity (RIN > 8.0) via spectrophotometry and bioanalyzer.
- Reverse Transcription: Using 500 ng - 1 µg total RNA, perform cDNA synthesis with a reverse transcriptase kit using oligo(dT) and/or random hexamer primers. Include a no-reverse transcriptase (-RT) control.
- qPCR Reaction Setup:
- Use a SYBR Green or TaqMan probe-based master mix.
- Primers: Design amplicons spanning an exon-exon junction to preclude genomic DNA amplification. Validate primer efficiency (90-110%).
- Reaction: 10 µL final volume: 5 µL master mix, 0.5 µL each primer (10 µM), 1 µL cDNA (diluted 1:10), 3 µL nuclease-free water.
- Run in technical triplicates.
- Thermocycling: Standard two-step protocol: 95°C for 3 min (initial denaturation); 40 cycles of 95°C for 10 sec (denaturation) and 60°C for 30 sec (annealing/extension).
- Data Analysis: Calculate ∆Ct values relative to a stable endogenous control (e.g., GAPDH, ACTB). Use the comparative ∆∆Ct method to determine fold-change differences between experimental groups. Statistical significance tested via t-test on ∆Ct values.
Western Blotting
Purpose: To detect and semi-quantify specific proteins and their post-translational modifications (PTMs) identified via proteomics. Detailed Protocol:
- Protein Extraction & Quantification: Lyse validation cohort cells/tissues in RIPA buffer with protease and phosphatase inhibitors. Clarify by centrifugation. Quantify protein concentration using a BCA assay.
- Gel Electrophoresis: Load 20-40 µg of total protein per lane onto a 4-20% gradient SDS-polyacrylamide gel. Include a pre-stained molecular weight marker. Run at constant voltage (120-150V) until the dye front reaches the bottom.
- Transfer: Perform wet or semi-dry transfer onto a PVDF or nitrocellulose membrane. Confirm transfer with Ponceau S staining.
- Blocking & Antibody Incubation:
- Block membrane in 5% non-fat milk or BSA in TBST for 1 hour at room temperature.
- Incubate with primary antibody (specific to target protein or PTM) diluted in blocking buffer overnight at 4°C.
- Wash 3 x 5 min with TBST.
- Incubate with appropriate HRP-conjugated secondary antibody for 1 hour at room temperature.
- Wash 3 x 5 min with TBST.
- Detection: Incubate membrane with chemiluminescent substrate (e.g., ECL). Image using a CCD-based imager within the linear detection range.
- Stripping & Re-probing: Strip membrane with mild stripping buffer and re-probe for a loading control (e.g., β-Actin, GAPDH).
- Densitometric Analysis: Use software (ImageJ, ImageLab) to quantify band intensity. Normalize target band intensity to its corresponding loading control. Report as relative protein expression.
Visualizing the Validation Workflow
Diagram 1: Orthogonal validation workflow from multi-omics discovery.
Diagram 2: Core principles of qPCR and Western Blot assays.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Orthogonal Validation Experiments
| Reagent / Material | Function in Validation | Key Consideration for Rigor |
|---|---|---|
| RNase Inhibitors | Prevents degradation of RNA during isolation for qPCR. | Essential for obtaining intact, high-quality RNA. |
| High-Capacity cDNA Reverse Transcription Kit | Converts mRNA to stable cDNA for qPCR templates. | Use kits with both random hexamers and oligo(dT) for comprehensive conversion. |
| TaqMan Gene Expression Assays | Sequence-specific primers & probe sets for target gene qPCR. | Offers high specificity; requires predesigned or validated assays. |
| SYBR Green Master Mix | Fluorescent dye that binds double-stranded DNA during qPCR. | More economical; requires post-run melt curve analysis to confirm specificity. |
| RIPA Lysis Buffer | Comprehensive buffer for total protein extraction for WB. | Must be supplemented with fresh protease/phosphatase inhibitors. |
| Phosphatase Inhibitor Cocktail | Preserves labile phosphorylation states during protein extraction. | Critical for validating phospho-proteomics findings. |
| HRP-Conjugated Secondary Antibodies | Enzymatically amplifies the primary antibody signal for WB detection. | Species-specific; choice depends on host of primary antibody. |
| Chemiluminescent Substrate (ECL) | Provides the luminescent signal for imaging WB bands. | Premium "clarity" or "forte" substrates offer wider linear dynamic range. |
| Validated Primary Antibodies | Binds specifically to the target protein or PTM of interest. | Most critical choice. Seek antibodies validated for WB, with cited applications in peer-reviewed literature. |
| Housekeeping Protein Antibodies (β-Actin, GAPDH, Vinculin) | Provides a loading control for WB normalization. | Must be verified for stable expression across all experimental conditions in the validation cohort. |
Functional Validation through siRNA/CRISPR Screens and Perturbation Experiments
In multi-omics research, integrating genomics, transcriptomics, proteomics, and metabolomics generates vast, correlative datasets. While powerful for hypothesis generation, these approaches often fall short of establishing causal, functional relationships between genes/proteins and phenotypic outcomes. Functional validation via targeted perturbation—specifically siRNA (loss-of-function) and CRISPR (loss- or gain-of-function) screens—provides the essential causal link. These experiments transform correlative multi-omics hits into validated targets and mechanistic insights, forming the critical bridge between observational data and biological understanding in the drug discovery pipeline.
Table 1: Key Perturbation Technologies for Functional Validation
| Technology | Mechanism | Primary Use | Duration of Effect | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| siRNA/shRNA | RNAi-mediated mRNA degradation | Loss-of-function (knockdown) | Transient (3-7 days) | Well-established, high-throughput compatible | Off-target effects, incomplete knockdown |
| CRISPR-Cas9 Knockout | DSB repair by error-prone NHEJ | Permanent loss-of-function | Stable | High specificity, permanent modification, multiplexable | Off-target edits, slower phenotype onset |
| CRISPRi (Interference) | dCas9 fused to repressive domains (e.g., KRAB) blocks transcription | Reversible loss-of-function | Stable while expressed | Reversible, minimal off-target transcriptional effects | Requires sustained dCas9 expression |
| CRISPRa (Activation) | dCas9 fused to activators (e.g., VPR, SAM) recruits transcriptional machinery | Gain-of-function | Stable while expressed | Targeted gene activation, multiplexable | Context-dependent activation efficiency |
Table 2: Quantitative Output from a Representative Genome-wide CRISPR Screen (Hypothetical Data)
| Gene Target | sgRNA Sequence (Example) | Pre-Screen Read Count | Post-Selection Read Count | Log2(Fold Change) | FDR-adjusted p-value | Interpretation |
|---|---|---|---|---|---|---|
| Essential Gene (e.g., PCNA) | GACCTCCAATCCAAGTCGAA | 452 | 12 | -5.23 | 1.2e-10 | Essential for proliferation |
| Validated Hit | CTAGCCTACGCCACCATAGA | 511 | 1250 | +1.29 | 3.5e-05 | Confers resistance to drug X |
| Negative Control | AACGTTGATTCGGCTCCGCG | 488 | 502 | +0.04 | 0.82 | Non-targeting control |
| Positive Control | GACTTCCAGCTCAACTACAA | 465 | 10 | -5.54 | 4.1e-11 | Essential gene control |
Detailed Experimental Protocols
Protocol 1: Arrayed siRNA Screen for Hit Validation
Objective: Validate candidate genes from a transcriptomics study in a specific phenotype (e.g., cell viability).
- Design: Select 3-4 independent siRNAs per target gene. Include non-targeting siRNA (negative control) and siRNA against an essential gene (positive control).
- Reverse Transfection:
- Dilute siRNA in an appropriate buffer (e.g., 1X siRNA buffer).
- Mix diluted siRNA with transfection reagent (e.g., Lipofectamine RNAiMAX) in Opti-MEM medium. Incubate 20 min.
- Seed cells onto siRNA-lipid complexes in 96- or 384-well plates.
- Incubation: Culture cells for 72-96 hours to allow for mRNA knockdown.
- Phenotypic Assay: Perform assay (e.g., CellTiter-Glo for viability, high-content imaging for morphology).
- Analysis: Normalize data to controls. Require ≥2 siRNAs producing concordant phenotypes for validation.
Protocol 2: Pooled CRISPR-Cas9 Knockout Screen
Objective: Identify genes essential for cell survival under a selective pressure.
- Library Design: Use a genome-scale sgRNA library (e.g., Brunello, ~4 sgRNAs/gene).
- Virus Production: Package lentiviral sgRNA library in HEK293T cells. Titrate to achieve low MOI (<0.3) for single sgRNA integration.
- Cell Infection & Selection: Infect target cells at a high representation (~500 cells/sgRNA). Select with puromycin for 3-5 days.
- Population Split & Selection: Split cells into treated (e.g., with drug) and untreated control arms. Culture for 14-21 days, maintaining representation.
- Genomic DNA Extraction & NGS Prep: Harvest cells. Extract gDNA. Amplify integrated sgRNA cassettes via PCR with indexed primers for multiplexing.
- Sequencing & Analysis: Sequence on an Illumina platform. Align reads to the library reference. Use MAGeCK or similar tools to calculate sgRNA depletion/enrichment and identify significantly perturbed genes.
Visualization of Workflows & Pathways
Title: Functional Validation Workflow from Multi-omics to Hit
Title: CRISPRi vs CRISPRa Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Resources for Perturbation Screens
| Category | Item | Function & Description |
|---|---|---|
| Libraries | Genome-wide sgRNA (e.g., Brunello, GeCKO) | Pre-designed, pooled libraries for CRISPR knockout screens. |
| siRNA libraries (e.g., ON-TARGETplus) | Pre-designed, sequence-verified siRNA sets for arrayed RNAi screens. | |
| Delivery Tools | Lentiviral Packaging Systems (psPAX2, pMD2.G) | Second/third-generation systems for safe, high-titer sgRNA/shrNA virus production. |
| Transfection Reagents (Lipofectamine RNAiMAX, X-tremeGENE) | Chemical reagents for efficient siRNA/plasmid delivery in arrayed formats. | |
| Electroporation Systems (Neon, Nucleofector) | Physical methods for high-efficiency delivery in hard-to-transfect cells. | |
| Enzymes & Cloning | Cas9 Nuclease (WT, HiFi), dCas9-KRAB/VPR | Engineered proteins for DNA cleavage or transcriptional modulation. |
| Restriction Enzymes & Ligases (BsmBI, T4 DNA Ligase) | For cloning sgRNAs into lentiviral backbone vectors (e.g., lentiGuide-puro). | |
| Selection & Detection | Puromycin, Blasticidin, Hygromycin B | Antibiotics for selecting cells successfully transduced with resistance-bearing vectors. |
| Cell Viability Assays (CellTiter-Glo, AlamarBlue) | Luminescent/fluorescent readouts for proliferation/cytotoxicity screens. | |
| Analysis Software | MAGeCK, CRISPResso2, pinAPL-py | Bioinformatic tools for identifying enriched/depleted sgRNAs and analyzing editing efficiency. |
Within the burgeoning field of multi-omics data analysis research, the integration of disparate, high-dimensional datasets—such as genomics, transcriptomics, proteomics, and metabolomics—is paramount for constructing a holistic view of biological systems and disease mechanisms. This integration is critical for researchers, scientists, and drug development professionals aiming to identify robust biomarkers and therapeutic targets. The efficacy of this research is heavily dependent on the computational tools chosen for data fusion and analysis. This guide provides a technical comparison of major integration tools, evaluating their performance, underlying methodologies, and suitability for specific use cases in multi-omics research.
Core Methodologies for Multi-Omics Integration
Integration tools generally fall into three methodological categories: early integration (concatenation-based), intermediate/late integration (model-based), and hybrid approaches. The choice of methodology impacts interpretability, scalability, and the ability to handle noise and batch effects.
Early Integration (Concatenation)
Raw or pre-processed datasets from multiple omics layers are merged into a single composite matrix prior to downstream analysis (e.g., PCA, clustering).
- Protocol: 1) Normalize and scale each omics dataset individually. 2) Perform horizontal (sample-wise) concatenation into a matrix of dimensions
[samples x (features_omic1 + features_omic2 + ...)]. 3) Apply dimensionality reduction or statistical modeling on the combined matrix. - Use Case: Suitable for a limited number of omics layers where the total feature count does not vastly exceed sample count.
Late Integration (Model-Based)
Analyses are performed on each omics dataset independently, and the results (e.g., clusters, latent factors) are integrated in a subsequent step.
- Protocol: 1) Apply unsupervised learning (e.g., NMF, clustering) to each omics dataset separately to obtain sample-wise patterns. 2) Use consensus methods or statistical frameworks to find agreement across the omics-specific patterns.
- Use Case: Effective when data types are heterogeneous or have different scales/technical noise profiles.
Intermediate Integration (Multi-View Learning)
Seeks a joint low-dimensional representation shared across all omics datasets simultaneously. This is the most common approach for advanced tools.
- Protocol: 1) Define an objective function that maximizes the correlation or covariance between latent factors of different omics datasets (e.g., CCA, PLS). 2) Optimize the model to find a set of components that explain the co-variation across all inputs. 3) Use these joint components for downstream biological inference.
- Use Case: Ideal for identifying shared signals across omics layers and for predictive modeling where one omics layer can inform another.
Performance Comparison of Major Tools
The following table summarizes the quantitative performance, strengths, and weaknesses of prominent multi-omics integration tools, based on recent benchmarking studies.
Table 1: Comparison of Major Multi-Omics Integration Tools
| Tool Name | Core Methodology | Primary Strength | Key Weakness | Optimal Use Case | Input Data Types |
|---|---|---|---|---|---|
| MOFA+ (Multi-Omics Factor Analysis) | Bayesian statistical framework for unsupervised integration. | Handles missing data natively; provides interpretable factors; excellent for population-scale data. | Computationally intensive for very large feature sets (>20k features/layer). | Identifying co-variation across omics in cohort studies (e.g., TCGA). | Any continuous or binary data (RNA-seq, methylation, somatic mutations). |
| Integrative NMF (iNMF) | Non-negative Matrix Factorization with joint factorization constraint. | Learns both shared and dataset-specific factors; good for high-dimensional data. | Requires parameter tuning (lambda, k); results can be sensitive to initialization. | Deconvolving cell types or states in single-cell multi-omics data. | scRNA-seq, scATAC-seq, CITE-seq (count matrices). |
| mixOmics | Multivariate statistical (PLS, CCA, DIABLO). | Extensive suite of methods; strong for supervised/classification tasks; excellent visualization. | Assumes linear relationships; performance degrades with high sparsity. | Predictive biomarker discovery and supervised classification (e.g., disease outcome). | All major omics types (requires matched samples). |
| LRAcluster | Low-Rank Approximation based clustering. | Fast, memory-efficient; effective for identifying multi-omic cancer subtypes. | Primarily a clustering tool; less focused on latent factor interpretation. | Unsupervised patient stratification/subtyping from >2 omics layers. | Matrix format (e.g., gene expression, copy number, methylation). |
| Seurat (v4+) | Canonical Correlation Analysis (CCA) & Reciprocal PCA (RPCA). | Industry standard for single-cell; robust workflow for cell alignment and label transfer. | Designed primarily for single-cell data; less generic for bulk omics. | Integrating multi-modal single-cell data or batch correction across scRNA-seq datasets. | scRNA-seq, scATAC-seq, spatial transcriptomics. |
Detailed Experimental Protocol: Benchmarking Integration Tools
A standard benchmarking protocol is crucial for evaluating tool performance in a multi-omics research context.
Protocol: Benchmarking Integration Tool Performance on a Reference Dataset (e.g., TCGA BRCA)
Data Acquisition & Preprocessing:
- Source: Download level 3 bulk RNA-seq (gene expression), DNA methylation (450k array), and copy number variation (CNV) data for matched samples from the TCGA-BRCA cohort via the UCSC Xena browser or TCGAbiolinks R package.
- Preprocessing: Filter lowly expressed genes (RNA-seq), remove cross-reactive probes (methylation), and segment CNV data. Perform quantile normalization and log2 transformation where appropriate. Retain only samples with data across all three modalities (N ~ 800).
Ground Truth Definition:
- Use the established PAM50 molecular subtypes (LumA, LumB, Her2, Basal, Normal-like) as the biological ground truth for evaluation.
Tool Execution:
- Apply each integration tool (MOFA+, mixOmics DIABLO, LRAcluster) according to its vignette.
- For MOFA+: Run with default parameters, extracting 10 factors. Use factors as features for downstream clustering.
- For mixOmics (DIABLO): Set up a supervised design to discriminate PAM50 subtypes, tuning the number of components via
perf(). - For LRAcluster: Input the three matrices and perform joint clustering with optimal rank selection.
Performance Evaluation Metrics:
- Clustering Concordance: Use Adjusted Rand Index (ARI) and Normalized Mutual Information (NMI) to compare tool-derived clusters to PAM50 subtypes.
- Runtime & Memory: Record peak memory usage and wall-clock time on a standard compute node (e.g., 8 cores, 32GB RAM).
- Biological Relevance: Perform enrichment analysis (GO, KEGG) on features weighted heavily in the key integrative components and compare to known breast cancer biology.
Visualizing Multi-Omics Integration Strategies
Diagram 1: Core Multi-Omics Data Integration Strategies
Diagram 2: MOFA+ Integration Model Workflow
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 2: Essential Research Reagents and Computational Resources for Multi-Omics Integration Studies
| Item | Function & Explanation |
|---|---|
| Reference Multi-Omics Datasets (e.g., TCGA, CPTAC, Human Cell Atlas) | Provide standardized, clinically annotated, matched multi-omics data for method development, benchmarking, and hypothesis generation. |
| High-Performance Computing (HPC) Cluster or Cloud Instance (e.g., AWS EC2, Google Cloud) | Essential for running memory-intensive and parallelizable integration algorithms on large-scale datasets (N > 1000 samples). |
| Conda/Bioconda Environment | A package manager for creating reproducible, isolated software environments containing specific versions of integration tools (R/Python) and their dependencies. |
| Singularity/Docker Container | Containerization technology that encapsulates an entire analysis pipeline, ensuring absolute reproducibility and portability across different computing systems. |
Benchmarking Workflow (e.g., SuPERR or custom Snakemake/Nextflow pipeline) |
Automated workflow to run multiple integration tools with consistent preprocessing and evaluation metrics, enabling fair comparison. |
Selecting the optimal integration tool for a multi-omics research project is contingent upon the biological question, data characteristics, and analytical goals. MOFA+ excels in exploratory, unsupervised discovery of latent factors across population data. mixOmics is a versatile toolkit ideal for supervised biomarker identification. For single-cell multi-omics, Seurat and iNMF are leaders. Researchers must weigh strengths in interpretability, handling of missing data, scalability, and supervised vs. unsupervised capabilities. A rigorous, protocol-driven benchmarking approach using standardized metrics is indispensable for validating tool performance within the specific context of one's research thesis on multi-omics data integration.
Evaluating the Biological Concordance and Novelty of Integrated Results
Within the broader thesis on Introduction to multi-omics data analysis research, a critical final step is the rigorous evaluation of the biological plausibility and novelty of the findings. This guide details the framework for assessing biological concordance (the agreement of new results with established knowledge) and novelty (the identification of previously unreported insights) in integrated multi-omics studies.
Quantitative Data on Multi-Omics Concordance & Novelty
The following table summarizes key metrics and statistical approaches for evaluating integrated results.
Table 1: Metrics for Evaluating Biological Concordance and Novelty
| Evaluation Dimension | Quantitative Metric / Method | Typical Value/Range (Benchmark) | Interpretation |
|---|---|---|---|
| Pathway Concordance | Overlap with known pathways (e.g., KEGG, Reactome) using hypergeometric test. | Adjusted p-value < 0.05 | Significant enrichment indicates high biological concordance with established mechanisms. |
| Network Concordance | Jaccard Index or Spearman correlation comparing inferred network with a gold-standard reference network. | Jaccard Index: 0.1-0.3 (highly variable by context) | Higher index suggests greater topological agreement with known interactions. |
| Novelty: Entity-Level | Percentage of key biomarkers (genes, proteins, metabolites) not previously associated with the phenotype/disease in major databases (e.g., DisGeNET, GWAS Catalog). | ~10-30% novel entities common in discovery studies. | High percentage may indicate a novel finding but requires robust validation. |
| Novelty: Relationship-Level | Number of predicted novel edges (interactions, regulations) in an integrated network not present in reference databases (e.g., STRING, OmniPath). | Varies widely; statistical significance assessed via permutation testing. | Novel edges suggest new mechanistic hypotheses. |
| Multi-Omic Concordance | Canonical Correlation Analysis (CCA) or DIABLO (mixOmics) between-omics block correlation. | CCA correlation > 0.7 indicates strong shared signal. | High correlation shows coherent biological signal across data layers. |
Experimental Protocols for Validation
Protocol 3.1: Orthogonal Validation of Novel Biomarkers via qRT-PCR
Purpose: To validate transcriptomic findings from an integrated analysis.
- Sample Preparation: Use the same biological samples (or replicates) from the original omics study.
- RNA Isolation: Extract total RNA using a column-based kit (e.g., RNeasy Mini Kit). Assess purity (A260/A280 ~1.9-2.1) and integrity (RIN > 7.0).
- cDNA Synthesis: Perform reverse transcription with 1 µg RNA using a High-Capacity cDNA Reverse Transcription Kit with random hexamers.
- qPCR Assay Design: Design primers for 3-5 novel gene targets and 2-3 reference genes (e.g., GAPDH, ACTB). Use a primer design tool (e.g., Primer-BLAST) for 80-150 bp amplicons.
- qPCR Reaction: Set up 20 µL reactions in triplicate using SYBR Green Master Mix, 10 ng cDNA, and 200 nM primers.
- Data Analysis: Calculate ∆Ct (Cttarget - Ctreference). Use the comparative ∆∆Ct method to determine relative expression changes between experimental groups. Statistical significance assessed via t-test (p < 0.05).
Protocol 3.2: Functional Validation of a Novel Pathway via CRISPR-Cas9 Knockout
Purpose: To test the causal role of a novel gene or pathway identified through integrated analysis.
- sgRNA Design & Cloning: Design two sgRNAs targeting exons of the novel gene of interest. Clone annealed oligos into a lentiviral CRISPR vector (e.g., lentiCRISPRv2).
- Virus Production: Co-transfect HEK293T cells with the sgRNA vector and packaging plasmids (psPAX2, pMD2.G). Harvest lentiviral supernatant at 48 and 72 hours.
- Cell Line Transduction: Transduce target cell line with virus in the presence of polybrene (8 µg/mL). Select with puromycin (1-5 µg/mL) for 5-7 days.
- Knockout Validation: Confirm gene knockout via Western blot (for protein) or Sanger sequencing of the target locus after PCR amplification.
- Phenotypic Assay: Perform a relevant functional assay (e.g., proliferation assay, migration assay, metabolite quantification via LC-MS) comparing knockout to wild-type cells.
- Rescue Experiment: Re-express a wild-type cDNA of the target gene in knockout cells to confirm phenotype reversal, establishing causality.
Visualization of Key Concepts
Title: Workflow for Evaluating Integrated Multi-Omics Results
Title: Example Integrated Pathway with Novel Elements
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Validation Experiments
| Reagent / Material | Provider Examples | Function in Evaluation |
|---|---|---|
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher, Bio-Rad | Converts RNA from multi-omics samples to cDNA for qPCR validation of transcriptomic hits. |
| SYBR Green qPCR Master Mix | Thermo Fisher, Qiagen, NEB | Enables quantitative, specific amplification of target sequences for biomarker validation. |
| lentiCRISPRv2 Vector | Addgene (deposited by Feng Zhang) | Lentiviral backbone for stable delivery of Cas9 and sgRNA for functional knockout experiments. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene | Essential for producing replication-incompetent lentiviral particles for gene editing. |
| Polybrene (Hexadimethrine bromide) | Sigma-Aldrich | Enhances lentiviral transduction efficiency in target cell lines. |
| Puromycin Dihydrochloride | Thermo Fisher, Sigma-Aldrich | Selective antibiotic for enriching cells successfully transduced with CRISPR vectors. |
| RIPA Lysis Buffer | Cell Signaling, Thermo Fisher | Efficiently extracts total protein from cells for Western blot validation of protein targets. |
| Pathway-Specific Small Molecule Inhibitors/Activators | Selleckchem, Tocris, MedChemExpress | Pharmacologically perturbs pathways of interest to test causality and concordance of network predictions. |
| LC-MS Grade Solvents (Acetonitrile, Methanol) | Fisher Chemical, Honeywell | Essential for high-sensitivity metabolomic validation assays following integrated discovery. |
Within the framework of multi-omics data analysis research, the ultimate challenge is the effective translation of computational predictions into clinically actionable insights. The translational pipeline, from high-dimensional omics data to patient impact, is fraught with biological complexity and technical validation hurdles. This guide outlines a systematic, evidence-based approach to rigorously assess the translational potential of multi-omics discoveries, focusing on the critical bridge between in silico prediction and in vivo relevance.
The Validation Pyramid: A Framework for Assessment
Translational assessment requires a multi-tiered validation strategy, moving from computational confidence to clinical proof-of-concept.
Table 1: The Multi-Tiered Translational Validation Framework
| Validation Tier | Primary Objective | Key Metrics & Outputs | Typical Experimental System |
|---|---|---|---|
| Tier 1: Computational Rigor | Ensure statistical robustness & biological plausibility of predictions. | False Discovery Rate (FDR), AUC-ROC, Pathway enrichment FDR, Network centrality scores. | In silico models, public repository data (TCGA, GTEx, PRIDE, etc.). |
| Tier 2: In Vitro Mechanistic | Confirm target existence, modulation, and direct phenotypic effect. | Protein expression (WB), mRNA fold-change (qPCR), CRISPR knockout viability, cellular assay IC50. | Immortalized cell lines, primary cells, 2D/3D cultures. |
| Tier 3: In Vivo Pharmacodynamic | Demonstrate target engagement and pathway modulation in a living organism. | Target occupancy assays, biomarker modulation in plasma/tissue, imaging (e.g., PET). | Mouse/rat models (xenograft, syngeneic, genetically engineered). |
| Tier 4: In Vivo Efficacy & Safety | Establish therapeutic effect and preliminary therapeutic index. | Tumor growth inhibition (TGI%), survival benefit (Kaplan-Meier), clinical pathology, histopathology. | Patient-derived xenograft (PDX) models, humanized mice, disease-relevant animal models. |
| Tier 5: Clinical Correlation | Link target/pathway to human disease biology and outcomes. | Association with patient survival, disease stage, treatment response in cohorts. | Retrospective analysis of clinical trial biopsies or well-annotated biobanks. |
Detailed Experimental Protocols for Key Validation Tiers
Protocol 3.1: Multi-Omics Target Prioritization & In Vitro Knockout Validation This protocol follows the identification of a candidate oncogene from integrated RNA-Seq and proteomics data.
- Computational Prioritization: From a list of differentially expressed genes/proteins, apply filters: (a) Log2FC > 2, (b) FDR < 0.01, (c) essentiality score (from DepMap CRISPR screens) < -0.5, (d) high network connectivity in a protein-protein interaction network.
- sgRNA Design & Lentiviral Production: Design three independent sgRNAs targeting exonic regions of the candidate gene using the Broad Institute's GPP Portal. Clone into a lentiviral vector (e.g., lentiCRISPRv2). Produce lentivirus in HEK293T cells via co-transfection with psPAX2 and pMD2.G packaging plasmids.
- Cell Line Transduction & Selection: Transduce target cancer cell line (e.g., A549) with viral supernatant in the presence of 8 µg/mL polybrene. After 48 hours, select transduced cells with 2 µg/mL puromycin for 7 days to generate a polyclonal knockout pool.
- Validation of Knockout & Phenotypic Assay: Confirm knockout via Western Blot (primary antibody specific to target) and T7 Endonuclease I assay. Assess phenotypic consequence using a CellTiter-Glo viability assay at 72h and 96h post-seeding. Compare growth to non-targeting sgRNA control.
Protocol 3.2: In Vivo Pharmacodynamic Assessment in a Xenograft Model This protocol assesses target engagement and pathway inhibition following treatment with a candidate inhibitory compound.
- Model Establishment: Subcutaneously inoculate 5x10^6 target cancer cells (with confirmed target expression) into the flanks of immunocompromised mice (e.g., NOD-scid IL2Rgammanull, NSG). Randomize mice into Vehicle and Treatment groups (n=8) when tumors reach ~150 mm³.
- Dosing & Monitoring: Administer compound or vehicle via oral gavage or IP injection at the predetermined maximum tolerated dose (MTD) schedule (e.g., QDx21). Measure tumor volumes bi-weekly using digital calipers (Volume = (Length x Width²)/2).
- Biomarker Collection & Analysis: At a predefined pharmacodynamic timepoint (e.g., 4h post-final dose), euthanize cohort subsets (n=4). Collect tumors and snap-freeze in liquid nitrogen. Perform:
- Western Blot: Analyze lysates for levels of phosphorylated target protein and downstream pathway effectors (e.g., p-ERK, p-AKT).
- qPCR: Quantify expression of transcriptional biomarkers indicative of pathway suppression.
- Immunohistochemistry (IHC): Stain formalin-fixed sections for a proliferation marker (Ki-67) and a cell death marker (cleaved Caspase-3).
Visualization of Core Concepts
Diagram 1: Sequential Flow of Translational Validation
Diagram 2: Example Targetable Signaling Pathway (PI3K-AKT-mTOR)
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagents for Translational Validation Experiments
| Reagent / Solution | Supplier Examples | Primary Function in Validation |
|---|---|---|
| CRISPR/Cas9 Knockout Kits | Horizon Discovery, Synthego, Thermo Fisher | Enables rapid genetic perturbation to test target necessity and sufficiency for phenotype. |
| Validated Antibodies for WB/IHC | Cell Signaling Technology, Abcam, CST | Critical for confirming protein expression, post-translational modifications (phosphorylation), and target engagement in vivo. |
| Phospho-Kinase Array Kits | R&D Systems, Proteome Profiler | Multiplexed screening to assess broad signaling pathway modulation upon target inhibition. |
| Patient-Derived Xenograft (PDX) Models | The Jackson Laboratory, Charles River, Champions Oncology | Preclinical models that better retain tumor heterogeneity and patient-specific drug responses. |
| Multiplex Immunoassay Panels (Luminex/MSD) | Luminex, Meso Scale Discovery | Quantify panels of soluble biomarkers (cytokines, phosphorylated proteins) from serum or tissue lysates with high sensitivity. |
| Next-Gen Sequencing Library Prep Kits | Illumina, Qiagen, New England Biolabs | For RNA-Seq or targeted sequencing to validate gene expression changes and discover resistance mechanisms. |
| Cell Viability/Proliferation Assays | Promega (CellTiter-Glo), Abcam (MTT) | Quantitative measurement of cellular health and proliferation following genetic or pharmacological intervention. |
| In Vivo Imaging Systems (IVIS) | PerkinElmer | Enables non-invasive tracking of tumor growth, metastasis, and reporter gene expression (e.g., luciferase) in live animals. |
Conclusion
Multi-omics data analysis represents a paradigm shift from a reductionist to a systems-level understanding of biology and disease. By mastering the foundational concepts, methodological workflows, troubleshooting techniques, and rigorous validation frameworks outlined here, researchers can move beyond single-layer observations to construct actionable, mechanistic models. The future of the field lies in the development of more dynamic, single-cell, and spatially-resolved multi-omics technologies, coupled with advanced AI-driven integration methods. For drug development, this holistic approach promises to deconvolve disease heterogeneity, identify robust composite biomarkers, and uncover novel, synergistic therapeutic targets, ultimately paving the way for more personalized and effective medicine. Success requires not only computational prowess but also close collaboration between bioinformaticians, biologists, and clinicians to ensure findings are both statistically sound and biologically meaningful.